










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Oftalmología
versión On-line ISSN 1561-3070
Rev Cubana Oftalmol vol.27 no.4 Ciudad de la Habana oct.-dic. 2014
PRESENTACIÓN DE CASO
Manifestaciones oftalmológicas de la epidermólisis bullosa
Ophthalmological manifestations of epidermolysis bulosa
MSc. Magela E. Díaz Rodríguez, MSc. Zaadia Pérez Parra, MSc. Elizabeth T. Escalona Leyva, MSc. Justo L. Noriega Martínez, MSc. Alexeide de la C. Castillo Pérez, MSc. Susana Márquez Márquez
Instituto Cubano de Oftalmología "Ramón Pando Ferrer". La Habana, Cuba.
RESUMEN
La epidermólisis bullosa es una genodermatosis, que comprende un grupo heterogéneo de enfermedades ampollares de la piel y las mucosas, localizadas en la membrana basal epidérmica y la capa basal del epitelio estratificado queratinizado o mucoso, con la consiguiente fragilidad de la piel y severidad variable en su presentación clínica. Con el objetivo de describir y mostrar los hallazgos oftalmológicos más comunes de esta afección, presentamos un caso cuyos síntomas iniciaron a los dos meses de edad con presencia de vesículas y ampollas localizadas en la región frontal, nasal, mejillas y caras laterales del cuello de la frente y las extremidades superiores e inferiores. Las manifestaciones oftalmológicas comenzaron a los seis meses de edad en ambos ojos con leucoma corneal cicatrizal total, simblefaron en 360º, que alcanzó región perilímbica. Todo lo anterior afectó su desarrollo visual y por consiguiente su desarrollo psicomotor. Esta afección requiere tratamiento multidisciplinario con especial atención a la superficie ocular externa para prevenir alteraciones que afecten la visión.
Palabras clave: epidermólisis bullosa, genodermatosis, cicatrización corneal.
ABSTRACT
Epidermolysis bulosa is defined as genodermatosis involving a heterogenous group of blistering diseases in the skin and the mucosas, located in the epidermal basal membrane and the basal layer of the stratified, keratinized or mucosal epithelium, with resulting fragility of the skin and variable severity in its clinical presentation. The most common eye findings of this disease were described and shown. The treatment of epidermolysis bulosa should be multidisciplinary, paying special attention to the outer ocular surface to prevent alterations that may affect the vision.
Key words: epidermolysis bulosa, genodermatosis, corneal scarring.
INTRODUCCIÓN
La epidermólisis bullosa (EB) representa una genodermatosis, que comprende un grupo heterogéneo de enfermedades ampollares de la piel y las mucosas, caracterizada por la presencia de ampollas y úlceras localizadas en la membrana basal epidérmica y la capa basal del epitelio estratificado queratinizado o mucoso, con la consiguiente fragilidad de la piel y severidad variable en su presentación clínica. Desde el punto de vista patológico, la epidermólisis bullosa se puede dividir en 4 grandes grupos: la epidermólisis simple o intraepidérmica (EBS), la epidermólisis de unión (EBU), la epidermólisis bullosa dermolítica o distrófica (EBD), y la mixta o síndrome de Kindler. Esta clasificación se basa en determinar a qué nivel de la epidermis se forman las bulas. Las herramientas diagnósticas más comúnmente empleadas para confirmar y clasificar el tipo de epidermolisis bullosa son: la microscopia electrónica y la utilización de la técnica de inmunomapeo antigénico. Ambas técnicas permiten determinar a qué nivel de la piel se encuentra la ampolla y/o separación: intraepidérmica (EBS), intralámina lúcida (EBU), sublámina densa (EBD) o mixta (síndrome de Kindler). La técnica de inmunomapeo antigénico se utiliza para valorar la presencia o ausencia de proteínas defectuosas en la unión dermoepidérmica y, de esa forma, subclasificar los tipos de estas genodermatosis. Los signos más obvios de esta enfermedad son la formación de vesículas y bulas dolorosas dentro de la piel y las membranas mucosas. Sin embargo, también existen alteraciones extracutáneas como vesículas y cicatrices en la boca, cicatrices en la conjuntiva y córnea, anomalías dentales, estenosis esofágicas, estenosis uretrales, fimosis y estenosis anales. La amplia gama de manifestaciones clínicas observadas en esta patología está relacionada con el gran número de defectos moleculares que presentan estos pacientes. Entre los dos tipos de enfermedades (adquirida y hereditaria) existen más de 20 fenotipos.1,2
Se estima que esta enfermedad afecta a uno de cada 17 000 nacidos vivos a nivel mundial, con una prevalencia de alrededor de 500 000 pacientes afectados en todo el mundo.3,4 En los Estados Unidos se ha estimado que existen 0,36 casos por cada millón de habitantes. En México no se conocen los datos epidemiológicos específicos de la enfermedad; sin embargo, la Asociación DEBRA México, A.C. (Dystrophic Epidermolysis Research Association) cuenta en su registro con alrededor de 269 casos de EB bien documentados desde 1998.4 Su etiopatogenia es desconocida, aunque se han descrito posibles causas entre las que se encuentran la existencia de adherencias amnióticas, alteraciones vasculares placentarias, infecciones intrauterinas, acción de teratógenos, defectos del cierre del tubo neural, rotura precoz de membranas y fuerzas de tensión, entre otras, y existen muchos casos sin asociación alguna ni causa probable.5
PRESENTACIÓN DEL CASO
Paciente de dos años de edad, del sexo femenino, con antecedentes de parto normal a las 38 semanas. No se recogen antecedentes prenatales, ni otros antecedentes patológicos familiares. Desde los dos meses de edad comenzaron manifestaciones dermatológicas como formación de vesículas y ampollas de la piel, que afectaron la región frontal, nasal, mejillas y caras laterales del cuello de la frente y las extremidades superiores e inferiores. A los seis meses de edad iniciaron las manifestaciones oftalmológicas en ambos ojos: leucoma corneal cicatrizal total y simblefaron en 360º, que alcanzó la región perilímbica. Todo lo anterior afectó su desarrollo visual y, por consiguiente, su desarrollo psicomotor.
La madre acudió a consulta y refirió mala visión y mancha blanquecina en ambos ojos. Al examen oftalmológico se constató, por esquiascopia, opacidad de los medios. No se pudo obtener la agudeza visual por la edad del paciente. En la biomicroscopia observamos en ambos ojos presencia de simblefaron en 360º, que alcanzó la región perilímbica, y leucoma corneal cicatrizal que impidió evaluar otras estructuras del segmento anterior (Fig. 1 y 2).

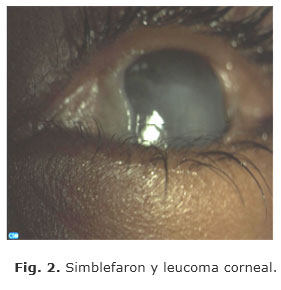
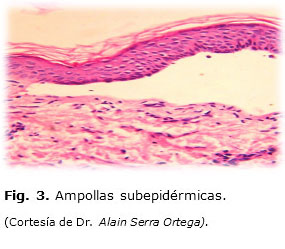
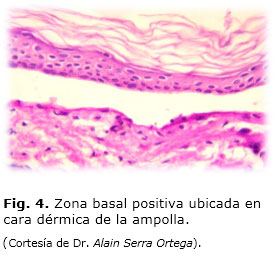
Se indicó ultrasonido de retina para evaluar las estructuras del segmento posterior y se informó retina aplicada, coroide, de grosor normal. No hiperreflectividad vítrea ni del área cristaliniana. Se observó hiperreflectividad en área corneal. La epidermólisis bullosa se confirmó por el estudio histológico convencional (hematoxilina/eosina) que mostró ampollas subepidérmicas (Fig. 3), y con la coloración de ácido periódico de Schiff (PAS) se pudo identificar que la zona basal positiva se ubicaba en la cara dérmica de la ampolla (Fig. 4). El informe de la biopsia indicó dermatitis vesicular subepidérmica con daño de la membrana basal e infiltrado celular mixto.
DISCUSIÓN
La epidermólisis bullosa distrófica recesiva (EBDR) es una genodermatosis rara, que puede afectar seriamente la visión del paciente. Existen varios reportes en la literatura sobre los hallazgos oculares de la epidermólisis bullosa. En Cuba encontramos el reporte de un paciente con epidermólisis bullosa asociado a aplasia cutis congénita y atresia pilórica (epidermólisis de la unión);6 sin embargo, no encontramos descripción de afectaciones oculares.En conclusión, en la epidermólisis bullosa la afectación ocular constituye una causa importante de morbilidad, ya que afecta gravemente la agudeza visual. En el caso presentado, las afectaciones que predominaron en la superficie ocular fueron el simblefaron y el leucoma corneal cicatrizal. Por eso es importante la atención multidisciplinaria para el tratamiento de estos pacientes, con un control oftalmológico más cercano que permita detectar y tratar las afecciones oculares que puedan surgir en estos casos.
REFERENCIAS BIBLIOGRÁFICAS
1. Stevenson M, Salas J, Páez JH, Rodríguez-García A. Prevalencia de manifestaciones oculares de la epidermólisis bullosa en México. Rev Mex Oftalmol. 2009 [citado 28 de marzo de 2014];83(6):[aprox 19 p.]. Disponible en: http://new.medigraphic.com/cgibin/resumen.cgi?IDREVISTA=87&IDARTICULO=23946&IDPUBLICACION=2447
2. Matsumoto Y, Dogru M, Tsubota K. Ocular surface findings in Hallopeau-Siemens subtype of dystrophic epidermolysis Bullosa: report of a case and literature review. Cornea. 2005 [citado 28 de marzo de 2014];24(4):[aprox 4 p.]. Disponible en: http://journals.lww.com/corneajrnl/pages/articleviewer.aspx?year=2005&issue=05000&article=00019&type=abstract
3. Featherstone C. Epidermolysis Bullosa: from fundamental molecular biology to clinical therapies. J Invest Dermatol. 2007 [citado 28 de marzo de 2014]; 127:[aprox 12 p.]. Disponible en: http://www.nature.com/jid/journal/v127/n2/full/5700731a.html
4. Salas-Alanis JC. Las epidermólisis bullosas. El proyecto DebRA. Med Cutan Iber Lat Am. 2007;35(4):165-6.
5. Pérez L, Urbina F, Roa J, Díaz Ch, Zambrano F. Aplasia cutis congénita: a propósito de cuatro casos. Rev Chil Pediatr. 2001;72(4):345-51.
6. Serra Ortega A, Sánchez Pérez OI, Clavel Isás D, Moreno Romo E. Epidermólisis bullosa, aplasia cutis congénita y atresia pilórica. Hallazgos histopatológicos en un neonato. Patología Autópsica. 2009;23:45-55.
7. Fine JD, Johnson LB, Weiner M, Stein A, Cash S, Deleoz J, et al. Eye involvement in inherited epidermolysis Bullosa: experience of the National Epidermolysis Bullosa Registry. Am J Ophthalmol. 2004 [citado 28 de marzo de 2014];138(2):[aprox 3 p.]. Disponible en: http://www.ajo.com/article/S0002-9394%2804%2900373-3/abstract
8. Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis Bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008 [citado 28 de marzo de 2014];58(6):[aprox 4 p.]. Disponible en: http://www.jaad.org/article/S0190-9622%2808%2900208-9/abstract
9. Khan AO. Corneal ulcer in a young child with autosomal recessive epidermolysis Bullosa. J Pediatr Ophthalmol Strabismus. 2006;43(6):370-2.
10. Tong L, Hodgkins PR, Denyer J, Brosnahan D, Harper J, Russell- Eggitt I, et al. The eye in epidermolysis Bullosa. Br J Ophthalmol. 1999 [citado 28 de marzo de 2014];83(3):[aprox 4p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1722980/
Recibido: 2 de julio de 2014.
Aprobado: 25 de agosto de 2014.
Dra. Magela E. Díaz Rodríguez. Instituto Cubano de Oftalmología "Ramón Pando Ferrer". Ave. 76 No. 3104 entre 31 y 41 Marianao, La Habana, Cuba. Correo electrónico: noraera@infomed.sld.cu

