










My SciELO
 Custom services
Custom servicesServices on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Oftalmología
Print version ISSN 0864-2176On-line version ISSN 1561-3070
Rev Cubana Oftalmol vol.30 no.3 Ciudad de la Habana July.-Sept. 2017
PRESENTACIÓN DE CASO
Hiperornitinemia con atrofia gyrata de coroides y retina
Hyperornithinemia with gyrate atrophy of the choroid and retina
Julio César González Gómez, Eliecer Pérez García, Odelaisys Hernández Echevarría, Daniel López Felipe, Yanelys Leal Delgado
Instituto Cubano de Oftalmología "Ramón Pando Ferrer". La Habana, Cuba.
RESUMEN
La atrofia gyrata de coroides y retina fue descrita por vez primera por Fuchs en el año 1896 como una entidad clínicamente definida. La deficiencia de la enzima ornitina delta aminotransferasa se hereda de forma autosómica recesiva; resulta en incremento plasmático de las concentraciones de ornitina y se asocia con atrofia gyrata de coroides y retina. Se presenta una paciente de 6 años de edad que es llevada a consulta, ya que en la escuela la maestra notaba mala visión de lejos. En un examen inicial del fondo de ojo el oftalmólogo observó cambios sugestivos de distrofia retiniana. En la oftalmoscopia binocular indirecta se encontraron extensas zonas confluentes de atrofia coroidea por fuera de las arcadas vasculares que respetaban el polo posterior; la mácula impresionaba normal. Se realizó un estudio de tomografía de coherencia óptica en dominio espectral en tomógrafo Spectralis que demostró la presencia de edema macular cistoide en ambos ojos. La determinación de niveles de ornitina en sangre arrojaron niveles muy elevados de este aminoácido (975 µmol/mL). Con todos estos hallazgos se llegó al diagnóstico de hiperornitinemia y atrofia gyrata de coroides y retina. Se indicó tratamiento dietético y vitamina B6 oral a pesar de que no se ha obtenido hasta el momento reducción significativa de los niveles de ornitina en plasma.
Palabras clave: atrofia gyrata; hiperornitinemia; distrofia de coroides.
ABSTRACT
Gyrate atrophy of the choroid and the retina was first described by Fuchs as a clinically defined condition in 1896. Human hereditary deficiency of ornithine aminotransferase activity is transmitted as an autosomal recessive trait and results in increased level of plasma ornithine and is associated with gyrate atrophy of the choroid and the retina. A 6-year-old girl was taken to the ophthalmologist’s because of her far poor vision detected by her teacher at the school. In the initial eye fundus examination the ophthalmologist observed some changes indicating retinal dystrophy. The indirect binocular funduscopy revealed extensive areas of choroidal atrophy outside the vascular archades respected the posterior pole whereas the macula impressed as normal. Cystoid macular edema was evident in both eyes according to the results of the optic coherence tomography performed with Spectralis tomograph. The aminoacid analysis revealed high serum ornithine level (975 µmol/mL). The clinical diagnosis of the patient was consistent with hyper-ornithinemia and gyrate atrophy of the choroid and the retina. She was treated with vitamin B6 and dietary supplementation but no significant reduction on her serum ornithine level was observed.
Key words: gyrate atrophy; hyperornithinemia; choroidal dystrophy.
INTRODUCCIÓN
La atrofia gyrata es una rara enfermedad de la coroides con una prevalencia de 1 en 50 000 habitantes en Finlandia. Se hereda de forma autosómica recesiva, aunque se han descrito algunos casos dominantes.1 El primer caso reportado de esta enfermedad fue descrito en el año 1888 por Jacobsohn como un ejemplo de “retinosis pigmentaria atípica”; no obstante, Cutler, en el año 1895, y Fuchs, en el año 1896, fueron los primeros en reconocer la enfermedad como una entidad clínicamente diferente.2 El trastorno bioquímico de esta enfermedad fue descrito inicialmente por Simell y Takki en el año 1973; consiste en una deficiencia de la enzima ornitina delta aminotransferasa (OAT) que resulta en incremento plasmático de las concentraciones de ornitina.3,4
Los síntomas iniciales incluyen mala visión nocturna y reducción de la visión periférica que usualmente comienza en la segunda o tercera década de la vida. Los cambios estructurales y funcionales se extienden desde la periferia hacia el centro, por lo que la disminución de la visión central ocurre tardíamente en la enfermedad. Miopía y cataratas subcapsulares posteriores son hallazgos frecuentes, así como pueden aparecer opacidades vítreas.1,5
PRESENTACIÓN DE CASO
Se trata de una paciente de 6 años de edad que es llevada a la consulta médica, pues en la escuela la maestra notaba mala visión de lejos. En un examen inicial del fondo de ojo el oftalmólogo observó cambios sugestivos de distrofia retiniana, por lo que es remitida al Servicio de Neuroftalmología. No se recogieron antecedentes personales ni familiares generales u oftalmológicos de interés. El examen inicial mostró una refracción ciclopéjica de -6,00 -1,00 x 180º para 20/20 en ojo derecho (OD) y -6,50 -1,00 x 35º para 20/25 en ojo izquierdo (OI). La visión de colores explorada con la prueba de Ishihara fue normal; los valores de tensión ocular con neumotonometría fueron de 13,7 mmHg en OD y 12,7 mmHg en OI. Los reflejos pupilares eran normales; la paciente se encontraba en ortotropía en posición primaria de la mirada y sin restricción en la motilidad ocular. El examen de los anejos y el segmento anterior en lámpara de hendidura no mostró alteraciones; los medios transparentes. En el fondo de ojo por oftalmoscopia binocular indirecta se encontraron discos ópticos de tamaño normal y ligeramente pálidos, discreto afinamiento vascular, extensas zonas confluentes de atrofia coroidea por fuera de las arcadas vasculares que respetaban el polo posterior; la mácula impresionaba normal (Fig. 1 y Fig. 2).
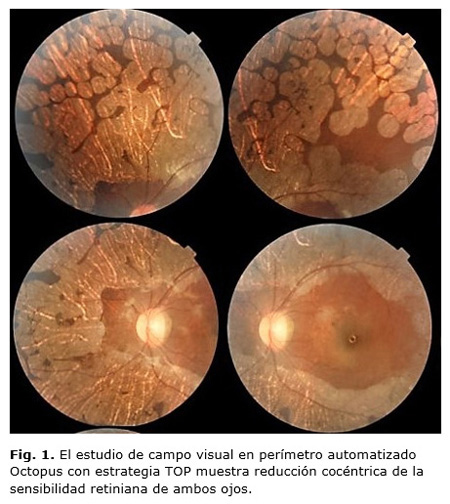
Se realizó un estudio de tomografía de coherencia óptica en dominio espectral en tomógrafo Spectralis que demostró la presencia de edema macular cistoide en ambos ojos (Fig. 3). El estudio de electromiografía fue normal. Se interconsultó con genética clínica y se realizó detección de niveles de ornitina en sangre que arrojaron niveles muy elevados de este aminoácido (975 µmol/mL). Con todos estos hallazgos se llegó al diagnóstico de déficit de ornitino delta aminotransferasa y atrofia gyrata de coroides. Se remitió a consulta de nutrición y se indicó tratamiento con vitamina B6 oral, a pesar de lo cual no se ha obtenido hasta el momento reducción significativa de los niveles de ornitina en plasma.
DISCUSIÓN
La enzima OAT es una enzima mitocondrial que utiliza al fosfato de piridoxina (vitamina B6) como cofactor que cataliza la interconversión de ornitina, glutamato y prolina. Esto resulta en anormalidades bioquímicas sistémicas que incluyen hiperornitinemia y reducción en plasma de lisina, glutamina, glutamato y creatina. Diferentes mutaciones han sido identificadas en el gen de la OAT en el cromosoma 10.6,7
Los cambios fundoscópicos comienzan en media periferia y la periferia con adelgazamiento y aspecto atrófico del epitelio pigmentario retiniano (EPR) debajo del cual los vasos coroideos pueden aparecer normales o escleróticos. Estas áreas son típicamente redondeadas inicialmente separadas, pero tienden -con la progresión lenta- a confluir centralmente y hacia la periferia. La progresión de la enfermedad lleva a acumulación de pigmentos, atrofia del el EPR y la coriocapilar y finalmente a la atrofia total de la coroides con exposición de la esclera. En estadios finales puede ser observado un anillo de coroides atrófica de la periferia hasta el polo posterior que usualmente respeta la mácula. Los vasos retinianos pueden aparecer normales al inicio o atenuados en estadios posteriores en que el nervio óptico puede aparecer pálido.1,5,8
Existen reportes de edema macular cistoide en pacientes con atrofia gyrata. La función visual varía considerablemente entre los casos y parece estar relacionada con la extensión del daño fundoscópico.9 El campo visual muestra constricción concéntrica como patrón más característico. No obstante, escotomas paracentrales y defectos anulares pueden presentarse en la evolución de la enfermedad. Finalmente si la fóvea se ve envuelta se observará un escotoma central.10
En estadios iniciales, el electrorretinograma estandarizado (ERGst) muestra solo anormalidades ligeras en la amplitud de la respuesta de conos y bastones, mientras que según progresa la enfermedad se deteriora la respuesta del ERGst y puede finalmente tornarse indetectable. La respuesta de bastones está afectada más severamente al inicio, pero luego -tanto la respuesta de bastones como la de conos- está severamente dañada. El electrooculograma (EOG) es normal o mínimamente disminuido en los inicios y marcadamente disminuido en estadios finales. El electromiograma (EMG) es con frecuencia anormal, aunque pocos pacientes aquejan debilidad muscular ligera. La biopsia de músculo muestra atrofia de fibras musculares tipo 2 con agregados tubulares. Anormalidades en el electrocardiograma (EKG) y el electroencefalograma (EEG) pueden verse en algunos pacientes.10
Una dieta restringida en arginina ha sido propuesta como forma de terapia en pacientes con atrofia gyrata, ya que la ornitina es producida a partir de otros aminoácidos, principalmente arginina. Algunos investigadores abogan por una dieta con restricción a niveles muy bajos de proteínas, incluyendo la eliminación casi total de la arginina con suplementos de aminoácidos esenciales.11 La administración oral de fosfato de piridoxina (vitamina B6) puede resultar en una reducción en plasma de oritina en algunos pacientes, mientras otros no responden a la vitamina B6.
Conflicto de intereses
El equipo de investigación declara no tener conflicto de intereses.
REFERENCIAS BIBLIOGRÁFICAS
1. Kim SJ, Lim DH, Kim JH, Kang SW. Gyrate atrophy of the choroid and retina diagnosed by ornithine-aminotransferase gene analysis: a case report. Korean J Ophthalmol. 2013;27(5):388-91.
2. Fuchs E. Ueber zwei der retinitis pigmentosa verwandte krankheiten (retinitis punctata albescens und atrophia gyrata choroideae et retinae). Arch Augenheilkd. 1896;32(9):111-6.
3. Moloney T, O'Hagan S, Lee L. Ultrawide-fild fundus photography of the fist reported case of gyrate atrophy from Australia. Clin Ophthalmol. 2014;8(7):1561-3.
4. Arshinoff SA, Leung K, Strube YNJ. Gyrate Atrophy. In: Duane's Clinical Ophthalmology. William Tasman M. Philadelphia: Lippincott Williams & Wilkins; 2005. p. 1456-64.
5. Valle D, Simell O. The hyperornithinemias. In: The metabolic and molecular bases of inherited disease. Scriver CR, Beaudet AL, Sly WS. New York: McGraw-Hill; 2001. p. 1875-95.
6. O'Donnell J, Cox D, Shows T. The ornithine amino-transferase gene is on human chromosome 10. Invest Ophthalmol Vis Sci. 1985;26:128.
7. Sergouniotis PI, Davidson AE, Lenassi E, Devery SR, Moore AT, Webster AR. Retinal structure, function and molecular pathologic features in Gyrate Atrophy. Ophthalmology. 2012;119(3):596-605.
8. Neha G, Pooja J, Supriya A, Basudeb G. Gyrate atrophy of the choroid and retina with cystoid macular edema and unilateral optic disc drusen. J Pediatr Ophthalmol Strab. 2015;52(12):64.
9. Sachet Prabhat S, Reema A, Rajesh P, Subramaniam B. First reported cases of gyrate atrophy of the choroid from Nepal. BMJ case reports. 2010;12(5):234-7.
10. Kaiser-Kupfer MI, Caruso RC, Valle D, Reed GF. Use of an argininerestricted diet to slow progression of visual loss in patients with gyrate atrophy. Arch Ophthalmol. 2004;122(3):982-4.
11. Weleber RG, Kennaway NG. Clinical trial of vitamin B6 for gyrate atrophy of the choroid and retina. Ophthalmology. 1981;88(14):316-24.
Recibido: 31 de enero de 2017.
Aprobado: 21 de marzo de 2017.
Julio César González Gómez. Instituto Cubano de Oftalmología "Ramón Pando Ferrer". Ave. 76 No. 3104 entre 31 y 41 Marianao, La Habana, Cuba. Correo electrónico: juliocgg@infomed.sld.cu


{kind=link}
{kind=link}