










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Protección Vegetal
versión impresa ISSN 1010-2752
Rev. Protección Veg. vol.28 no.3 La Habana sep.-dic. 2013
COMUNICACIÓN CORTA
Optimización de la amplificación por PCR del gen de la endoglucanasa de Ralstonia solanacearum
Optimization of the amplification by PCR of the endoglucanase gene in Ralstonia solanacearum (Smith) Yabuuchi
Eber NaranjoI, Adriana OteroII, Yamila Martínez-ZubiaurI
IDirección de Sanidad Vegetal. Centro Nacional de Sanidad Agropecuaria (CENSA). Apartado 10, San José de Las Lajas, CP 32 700, Mayabeque, Cuba. Correo electrónico: yamila@censa.edu.cu.
IIFacultad de Biología. Universidad de La Habana. Calle 25, No. 455, Vedado, CP 10400. La Habana, Cuba.
RESUMEN
El esquema de clasificación actual del agente etiológico de la marchitez bacteriana (Ralstonia solanacearum) se basa en el análisis de la secuencia del gen de la endoglucanasa de esta bacteria. El objetivo de este estudio fue optimizar la amplificación por PCR de este gen. Las condiciones de PCR fueron predeterminadas in silico con el empleo del programa OLIGO versión 7.53. Se estimó el efecto de la concentración celular en la eficiencia de la amplificación con diluciones seriadas de suspensiones bacterianas de R. solanacearum. Se evaluó un protocolo de PCR de dos pasos con el empleo del kit comercial GoTaq® DNA Polimerasa para los ensayos in vitro. La concentración celular fue un factor crítico para la amplificación con un valor óptimo de 108 UFC.ml-1. En el protocolo estandarizado se redujeron los tiempos en los ciclos de desnaturalización inicial y extensión final y se combinaron las etapas de hibridación y la extensión en un solo paso, por lo que resultó un protocolo más corto que el original. El análisis filogenético de la secuencia de este gen, permitirá estudiar la diversidad de los aislados cubanos de R. solanacearum, establecer relaciones con aislados de otras regiones geográficas y perfeccionar las estrategias de manejo de la enfermedad.
Palabras clave: marchitez bacteriana, in silico, in vitro.
ABSTRACT
The current classification scheme of the causal agent of the bacterial wilt Ralstonia solanacearum (Smith) Yabuuchi is based on the analysis of the endoglucanase gene sequence of the bacterium. The objective of this paper was to optimize the PCR amplification of this gene. PCR parameters were determined using the OLIGO software version 7.53 for in silico analyses. The effect of cell concentration on amplification efficiency was assessed by using serial dilutions of R. solanacearum suspensions. For the in vitro assays, a two step-PCR protocol was evaluated with the GoTaq® DNA Polimerasa kit. The cell concentration was a critical factor on amplification efficiency with an optimal value of 108 CFU.ml-1. Time was reduced for the initial denaturalization and final extension phases and hybridization and extension were combined in a single step resulting in a 57 minute shorter-PCR program than the original protocol. The phylogenetic analysis of this gene sequence will allow performing diversity studies of Cuban isolates of R. solanacearum, establishing comparative relationships with isolates of other geographical locations and updating the disease management strategies.
Key words: Bacterial wilt, in silico, in vitro.
El complejo de especies de Ralstonia solanacearum es el agente causal de la marchitez bacteriana, enfermedad que se encuentra diseminada a nivel mundial y que afecta a 54 familias del reino vegetal (1). Se conoce que este patógeno está ampliamente distribuido en el continente Americano, afecta a países con los que Cuba mantiene relaciones de intercambio, al mismo tiempo que las condiciones climáticas del país propician su desarrollo y supervivencia (2). Para Cuba, es una plaga de importancia económica con poca diseminación y bajo control con acciones legales de cuarentena y se cuentan con técnicas sensibles y específicas para su detección (2), sin embargo; se hace necesario disponer de herramientas para realizar estudios de epidemiología y variabilidad molecular, relaciones filogenéticas, distribución y procedencia geográfica de los aislados obtenidos, los que fortalecerían el sistema de diagnóstico del patógeno en Cuba.
El análisis filogenético de la secuencia del gen de la endoglucanasa (egl) demostró ser una herramienta útil, pues permite establecer un sistema de clasificación a nivel intraespecífico en correlación con el origen geográfico primario de los aislados (3, 4, 5).
En este sentido el objetivo de este trabajo fue optimizar la amplificación por PCR del gen de la endoglucansa de R. solanacearum, con el uso de herramientas bioinformáticas y convencionales, a fin de explotar su utilización en ulteriores análisis de filogenia y de epidemiología molecular de los aislados de R. solanacearum que se intercepten en el país.
En el trabajo se emplearon tres aislados de R. solanacearum pertenecientes al cepario del Laboratorio Central de Cuarentena Vegetal (LCCV) y el ADN total de un aislado obtenido en el Laboratorio de Bacteriología Vegetal del CENSA (Tabla 1). Se utilizaron suspensiones bacterianas en solución salina (NaCl al 0,85% en H2O destilada estéril) preparadas a una concentración de 109 UFC.ml-1 (DO540nm = 1,0) a partir de colonias puras en placas de agar en el LCCV. Las suspensiones se inactivaron por calor durante 10 min a 100oC y se realizaron 3 diluciones a razón de 1/10 por aislado.

La secuencia completa del gen egl obtenida de la base de datos del GenBank (No. de acceso M84922), se analizó en formato FASTA con el programa OLIGO versión 7.53 (6) y los iniciadores EndoF (5´-ATGCATGCCGCTGGTCGCCGC-3´) y EndoR (5´- GCGTTGCCCGGCACGAACACC -3´) (3) fueron anotados en la secuencia molde.
Para el análisis in silico se evaluaron los siguientes parámetros: 0,25 µM de concentración de iniciadores, 50 mM de concentración de sodio y 1,5 mM de concentración de magnesio. A partir de los parámetros óptimos de la reacción obtenidos en el análisis con el programa OLIGO 7.53 se establecieron las condiciones a emplear in vitro.
Para determinar el efecto de la concentración durante el proceso de optimización de la técnica se empleó el protocolo original descrito por Fegan y Prior (3) y se utilizó como molde, 1 µl de las suspensiones puras y diluidas 1/10 de cada uno de los aislados. El uso de las diluciones puras como molde mostró resultados no reproducibles, con la frecuente obtención de bandas inespecíficas y para algunos casos la inhibición total de la PCR. Cuando fueron empleadas las diluciones 1/10 (aproximadamente 108 UFC.ml-1) se obtuvo una buena intensidad de banda y se eliminaron los productos no deseados (Figura 1) por lo que éstas fueron empleadas para la optimización del protocolo.
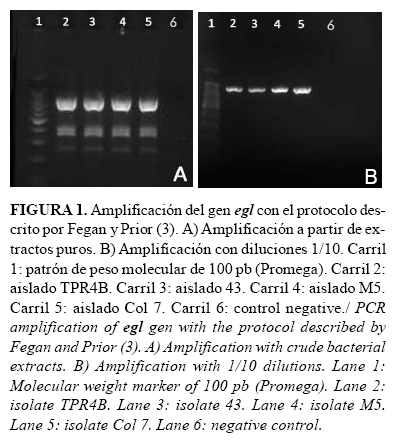
Los parámetros de la reacción in silico determinados con el programa se muestran en la Tabla 2. Los elevados valores de temperatura media de disociación para los iniciadores (Tm >70oC) sugirieron la aplicación de un protocolo de amplificación de dos pasos con las temperaturas de 95oC para la desnaturalización y entre 68-70oC para la hibridación y extensión simultánea. El tiempo medio de hibridación calculado por el programa para los iniciadores fue de 22,7 segundos. Está descrito que un minuto es suficiente para la amplificación de un fragmento de 1kb (5) por lo que fueron empleados 90 segundos para la fase de hibridación-extensión.
La mezcla de PCR del protocolo resultante estuvo compuesta por tampón de PCR al 5% facilitado por el fabricante (5X Green GoTaq® Reaction Buffer), 200 µM de cada dNTP; 1,25 U de la GoTaq® DNA Polimerasa (5 U/µl) y 0,25 µM de cada iniciador. El programa de amplificación de dos pasos constó de un ciclo inicial de desnaturalización a 95oC (2 min), seguido por 30 ciclos de: 95oC (1 min) y se ensayó con temperaturas de 68 y 70oC (90 seg) en un ciclo de hibridación y extensión simultáneas. El programa concluyó con un ciclo final de extensión de 72oC (5 min).
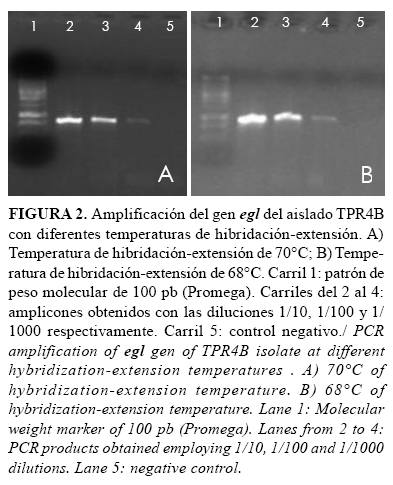
Se emplearon tres diluciones seriadas (1/10, 1/100 y 1/1000) del aislado TPR4B con el protocolo optimizado para observar el efecto de la temperatura de hibridación-extensión en la concentración de producto de PCR. Se obtuvieron bandas con buena resolución en las tres diluciones seriadas para ambas temperaturas (Figura 2). Sin embargo; se observó una mayor concentración del producto sin la presencia de productos inespecíficos en la evaluación del ensayo a 68°C, por lo que se determinaron como óptimas las condiciones empleadas en el mismo.
La concentración celular constituyó un factor crítico en la eficiencia de la amplificación, pues la obtención de resultados no reproducibles con las suspensiones puras hizo necesario el uso de las diluciones para evitar la presencia de productos inespecíficos. Es conocido que las suspensiones celulares y el exceso de ADN pueden ejercer un efecto inhibidor sobre la reacción de PCR. El exceso de ADN en el tubo de reacción puede dar lugar a uniones inespecíficas de los cebadores, aumento de la temperatura de desnaturalización (7) o a la inhibición de la ADN polimerasa por su unión a los polisacáridos ácidos de alto peso molecular de las paredes celulares (8), entre otros.
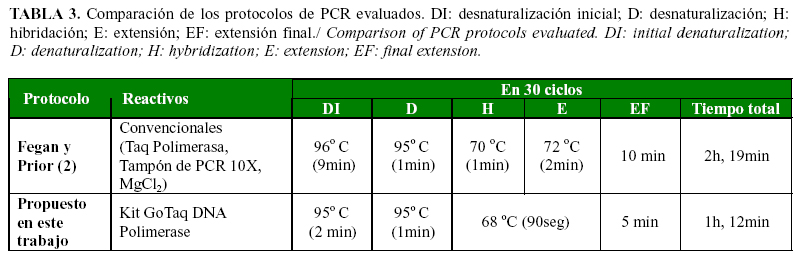
El empleo del programa OLIGO versión 7.53 permitió determinar in silico los parámetros de la reacción, con un considerable ahorro de reactivos durante el proceso de estandarización. Por otra parte, también influyó el empleo de un kit comercial en la mezcla de PCR. Estos formulados se usan con frecuencia para la amplificación de fragmentos con un elevado porcentaje de G+C pues contienen aditivos desestabilizadores de cadenas que introducen una astringencia adicional a la reacción (5) y permiten reducir el tiempo y los valores de temperatura empleados en cada ciclo. Tales variaciones se observan al comparar los tiempos y las temperaturas de las fases de desnaturalización inicial, hibridación y extensión final de los protocolos evaluados en este trabajo (Tabla 3).
Similares modificaciones fueron realizadas por otros autores para la amplificación del gen gyrB del género Xanthomonas (9, 10), en los cuales el uso del kit Long PCR Enzyme mix (Fermentas) permitió emplear una temperatura de hibridación 10oC menor a la temperatura óptima de hibridación de los iniciadores.
La metodología optimizada facilitará los estudios de caracterización de aislados de Ralstonia solanacearum, pues permitirá incrementar y simplificar el número de análisis para la detección de la variabilidad molecular, distribución geográfica y obtener una mayor información sobre los posibles orígenes de los aislados interceptados en las instancias de la cuarentena nacional.
El análisis de la secuencia nucleotídica del gen egl obtenido con este protocolo permitirá establecer relaciones filogenéticas entre los aislados que se detecten en el país e inferir la evolución de las poblaciones genéticas y sus patrones de variación, explotando la metodología como herramienta útil para conocer la evolución genética de poblaciones de patógenos exóticos y re-emergentes (11).
REFERENCIAS
1. Cellier G, Prior P. Deciphering phenotypic diversity of Ralstonia solanacearum strains pathogenic to potato. Phytopathology. 2010;100:1250-1261.
2. Iglesia A, Alvarez E, Martínez Y, García A. Diagnóstico molecular de la Marchitez Bacteriana. Rev Protección Veg. 2008;23(2):75-79.
3. Fegan M, Prior P. How complex is the Ralstonia solanacearum species complex? En: Allen C, Prior P y Hayward AC editors. Bacterial wilt disease and the Ralstonia solanacearum species complex. The American Phytopathology Society Press, St. Paul, MN 2005: 449-461.
4. Lemessa F, Zeller W, Negeri D. Genetic diversity among strains of Ralstonia solanacearum from Ethiopia assessed by repetitive sequence-based polymerase chain reaction (rep-PCR). EJAST. 2010;1(1):17-26.
5. Xu J, Pan ZC, Prior P, Xu JS, Zhang Z, Zhang H, et al. Genetic diversity of Ralstonia solanacearum strains from China. Eur J Plant Pathol. 2009;125:641-653.
6. Rychlik W. Ed. Yuryev A.; OLIGO 7 Primer Analysis Software, in Methods in Molecular Biology Vol. 402: PCR Primer Design; Humana Press Inc., Totowa, NJ. 2007:35-59.
7. Altshuler ML. PCR troubleshooting the essential guide. Editorial Castier Academic Press, Wymondham, U.K. 2008: 51-52.
8. Dittapongpitch V, Surat S. Detection of Ralstonia solanacearum in soil and weeds from commercial tomato fields using immunocapture and the polymerase chain reaction. Journal of Phytopathology. 2003;151:239-246.
9. Parkinson N, Cowie C, Heeney J, Stead D. Phylogenetic analysis of Xanthomonas species by comparison of partial gyrase B gene sequences. International Journal of Systematic and Evolutionary Microbiology. 2007;57:2881-2887.
10.Parkinson N, Cowie C, Heeney J, Stead D. Phylogenetic structure of Xanthomonas determined by comparison of gyrB sequences. International Journal of Systematic and Evolutionary Microbiology. 2009;59:264-274.
11.Grünwald NJ, Goss EM. Evolution and Population Genetics of Exotic and ReEmerging Pathogens: Novel Tools and Approaches Annu. Rev Phytopathol. 2011;49:249-67.
Recibido: 26-10-2012.
Aceptado: 5-4-2013.

{kind=link}
{kind=link}