










My SciELO
 Custom services
Custom servicesServices on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Archivo Médico de Camagüey
On-line version ISSN 1025-0255
AMC vol.5 no.5 Camagüey Set.-Oct. 2001
CASOS CLÍNICOS
Esclerosis sistémica progresiva
Progressive Systemic Sclerosis
Dr. Rafael Pila Pérez; Dr. Rafael Pila Peláez; Dra. Carmen Guerra Rodríguez; Rita Díaz Leal; Dra Margarita Pila Peláez; Kafui Twaku Tamakloe
Hospital Provincial Manuel Ascunce Domenech. Camagüey, Cuba.
RESUMEN
Se presenta el caso de una niña de 8 años con esclerosis sistémica progresiva, se señala si se trata de una forma sistémica infantil, de la cual hay muy pocos casos publicados y a favor de lo cual hay signos de afectación general; o si se trata de una forma localizada tipo morfea ya estabilizada, y que podría explicar la totalidad de la clínica. Se exponen las características de nuestro caso, así como la conducta asociada en su estudio y terapéutica.
DeCS: ESCLERODERMIA SISTÉMICA/terapia.
ABSTRACT
The case of an 8 year-old girl with Progressive Systemic Sclerosis is presented, it is pointed out if you if is an infantile systemic form, of wich there are very few published cases and in favour of which there are signs of generall affectation, or if it is a localized form, type morhpea, already estabilized, and which could explain the clinical manifestations. The charactistics of our case are exposed , as well as the measures associated with is study and treatment .
DeCS: SCLERODERMA, SYSTEMIC/therapy.
INTRODUCCIÓN
La esclerosis sistémica progresiva (ESP) es un trastorno que afecta múltiples órganos y sistemas, y se caracteriza por grados diversos de cambios en vasos, fibrosis e inflamación de la piel y órganos internos.1 De manera característica la enfermedad pasa por varias fases de inflamación y atrofia.2 La duración de estas fases varía en diferentes órganos. Su incidencia es tres veces mayor en el sexo femenino y la edad de aparición está entre los 22 y 55 años, es muy rara su presentación antes de los 25 años y excepcional en la infancia.3
Hay múltiples clasificaciones de la ESP como son la de Winkelman,4 Giordano,5 Rodnan.6 Barnett.7 Leroy8 y la de Siegel9 que es para nosotros la más completa.
La esclerodermia presenta diferentes puntos de vista clínicos, de acuerdo a los autores y a los diferentes países, nosotros señalamos el espectro más utilizado en la clínica. El motivo de este trabajo es la presentación del caso de una niña de 8 años, con manifestaciones que ponen en duda si se trata o no de una afectación sistémica, pues dada la edad la colocaría entre los pocos casos descritos de ESP infantil.
PRESENTACIÓN DEL CASO
Niña de 8 años de edad, desnutrida, del panículo adiposo, talla 1,27 cms, peso 20 Kg y un tercer percentil refiere que desde los 7 años se quejaba de dolores articulares que le impedían realizar las labores habituales como escribir en el colegio y presenta fiebre de 39ºC por lo que se ingresa.
Al interrogatorio: señala que desde hace ocho meses o más nota adormecimiento de ambas manos y pies, calambres nocturnos y dificultad para abrir y cerrar las manos, así como para la marcha. No presentaba antecedentes personales ni familiares de interés.
Examen Físico: Fiebre de 38ºC, palidez de piel y mucosas. La piel endurecida, seca, lisa adherida a planos profundos, con manchas hipocrómicas en manos, flexuras de los codos y rodillas, zonas de retracción al nivel de los codos, rodillas y región cervical de aspecto escamoso.
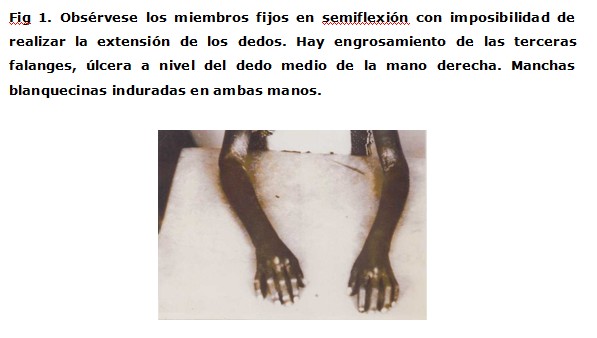
Las manos presentaban dedos afilados con ulceración a nivel del dedo medio de la mano derecha, dificultad para movilizar los miembros superiores e inferiores por ligera contractura en flexión, dificultad a la marcha (figura 1 y 2 ). Dificultad para abrir la boca, nariz afilada, pérdida de los pliegues faciales, lo que daba una facie de Tapir(figura 3). El pelo era escaso y quebradizo.
El aparato respiratorio y cardiovascular no presentaba alteraciones, pero se palpaba hepatomegalia de 3 cm que rebasaba el borde costal, algo dolorosa y lisa, sin detectar otras alteraciones al examen físico.
Estudio analítico:
Hb 8,5 g/l, leucocitos 7,500 x 10-9 /l con diferencial normal, Vsg 50 mm/hora; glicemia, creatinina, iones, enzimas, TASO, factor reumatoide, células LE (3), VDRL, Proteína C reactiva, DNA, crioglobulinas, anticuerpos, anticuerpos antimúsculo liso, complemento, de pruebas de Coombs directa e indirecta, conteo de reticulocitos normales. Constantes, Corpusculares: anemia ferropénica. Conteo de Addis: leucocituria, urocultivo: + de 100 000 colonias de E. coli sensible a tratamiento con ciprofloxacina. Radiografía de tórax: Survey óseo: desmineralización del carpo y extremos proximales y distales metacarpianos con lesiones de aspecto de "punta de lápiz", hiperdistención de las muñecas y de flexión de los dedos y ambos codos. Ecografía abdominal: hepatomegalia de 3 cm, sin otras alteraciones. Radiografía de esófago, estómago y duodeno: normal, no se pudo realizar manometría esofágica. Electroforesis de proteínas: hiperganma globulinemia (1,98%), radiografía de la boca: engrosamiento de la membrana periodontal. HLA: negativo.
Biopsia de Piel: Le epidermis no muestra alteraciones y la dermis aparece aumentada de espesor francamente colagenizado, con haces gruesos y empaquetados. No se advierten folículos pilosos ni infiltrados inflamatorios perceptibles. Solo se observan algunas glándulas sudoríparas comprimidads por el tejido conectivo colágeno. (Lesiones compatibles con esclerodermia localizada tardía residual).
Biopsia de Músculo: Normal, ECG: Normal, ecocardiografía: normal, electromiografía: Normal, pruebas funcionales respiratorias: normales. La paciente mejoró de su anemia con dieta y sales de hierro, la infección urinaria se resolvió con ciprofloxacina y las manifestaciones dermatológicas y articulares mejoraron ostensiblemente con el empleo de D-penicilamina a las dosis de 500 mgs diarios.
Aumentó durante su ingreso 7kg, mejoró su movilidad tanto en miembros superiores como inferiores pudiendo deambular sin dificultad, las lesiones de piel oscurecieron. Durante tres años hemos seguido la evolución de esta paciente con controles bimensuales, después semestrales. No ha aparecido ningún otro síntoma clínico y su desarrollo pondoestatural ha mejorado grandemente. Cuando ha tenido dolores o molestias hemos utilizado la colchina 0.5mg tres veces al día en ciclos de 3 a 4 semanas, recuperándose de esta molestia en varias semanas.
COMENTARIOS
La esclerodermia en bandas, sin afectación sistémica, es frecuente en el sexo femenino y puede comenzar en edades juveniles. La anatomía patológica pasa por dos estadíos, pero en la mayoría de los cortes es frecuente que se entremezclen zonas en los dos estadíos.10
La ESP es excepcional en edades infantiles. La anatomía patológica es muy parecida a la que se presenta en las formas localizadas, pero aquí la oclusión vascular es más intensa y la afección es mayor .La piel se indura y adhiere más a las estructuras proximales, dificultando la motilidad, y en estas formas está afectado el músculo estriado que se atrofia de forma universal, dando signos en aparato locomotor, corazón, aparato digestivo, y sistema nervioso.3-9
El pulmón ofrece afectación intersticial que puede llegar a la forma en "panal de abeja"y las lesiones renales pueden ser similares a las de los LES o a los de la hipertensión maligna (11), a veces se asocia a la cirrosis biliar primaria o puede producir trastornos hepáticos que hacen pensar en alteración de este órgano.
La mayoría de los casos tienen intensa afectación osteoarticular y de ellos la tercera parte pueden comenzar antes de tener lesiones en piel, con poliartralgias, fundamentalmente a nivel interfalángico (12). El 90% de los enfermos presenta el fenómeno de Raymaud durante dos años como único signo. El 95% esclerodactilia, el 89% calcinosis y el 30% trastornos pigmentarios.13
Desde el punto de vista inmunológico, el 50% tiene aumento de las globulinas gamma; el 60% anticuerpos antinucleares; el 50% anticuerpos intermúsculo liso y contra la piel, esto en cuanto a anomalías relacionadas con la formación de anticuerpos.2 Genéticamente se asocia con HLA B9 y B8 y en el síndrome de CREST al B27.14
El pronóstico de esta enfermedad es muy variable en su forma sistémica, pues hay formas graves con una supervivencia menor a los 10 años, hay otros de larga evolución que cursan con exacerbación a veces tardías con tratamiento sintomático con vasodilatadores y bloqueadores alfadrenérgicos para el fenómeno de Raynaud,8 los antinflamatorios, y una serie de fármacos y terapéuticas que están en investigación, tales como: inmunosupresores, azatropina, clorambucil, metrotexate, calcitriol, estrógenos, trasplante autólogo, plasmaféresis, tocoretinate local.15,18
El caso que nos ocupa nos deja serias interrogantes, pues no sabemos si catalogarlo como una ESP o una esclerodermia localizada. En un niña de 8 años donde la forma sistémica es sumamente rara hasta el punto que el número de casos publicados es mínimo, pero donde la forma localizada puede presentarse con mayor frecuencia.
La paciente no presentó fenómeno de Raynaud evidente, pero si alteraciones osteoarticulares con imposibilidad para mover los codos; fijación en las muñecas y en flexión de los dedos y codos, así como la imposibilidad de la marcha. Radiológicamente hay desmineralización importante del carpo, metacarpianos y de cavidad bucal. Todo ello precediendo a la aparición de las manchas en la piel, y señalando cierta atrofia de manos y debilidad muscular, todo lo cual es más específico de las formas sistémicas que de las localizadas.
La única manifestación visceral reportada fue la hepatomegalia. La alteración genitourinaria fue una infección por E. coli, hallazgo que explicaría la fiebre de larga fecha y que necesitó de quinolonas para su erradicación.
El estudio analítico practicado, todo ello aún con ANA, antimúsculo liso negativo, nos permiten pensar en alteración sistémica, máxime cuando estas pruebas se normalizan sólo con terapéutica esteroidea, volviendo a alterarse al suprimirla, como no apreciamos en este caso.19 Estas alteraciones no se describen en las formas localizadas, además los controles a esta paciente no han demostrado cambios en los mismos ni aparición de ningún otro signo o síntoma. En personas tratadas con D-penicilamina, disminuye la concentración de enlaces cruzados almidínicos reducibles en la piel, y hay una disfunción en la síntesis de la colagenasa.1,8,9 Los enfermos con esclerodermia que recibieron D-penicilamina apreciaron reblandecimiento de las lesiones cutáneas.
Las pruebas actuales no indican que grandes dosis produzcan mejorías visceral uniforme y por esta razón es razonable emplear dosis en el orden de 250 a 500 mgs al día, como la empleamos nosotros en este caso, para llevar al mínimo los efectos tóxicos y aún así obtener los beneficios cutáneos en personas con lesiones extensas de la piel.16,17 El otro fármaco empleado en este caso fue la colchicina, la cual se piensa que estimula la actividad de colagenasa e ihnibe la biosíntesis de colágena al evitar la secreción de colágena por célula.
En fechas recientes se ha sugerido que la actividad de colagenasa en la piel puede disminuir en la esclerodermia.20 Este fármaco fue urtilizado por nosotros a razón de 0.5mg tres veces al día, cinco días a la semana en ciclos de tres a cuatro semanas, alternando con la D-penicilamina.
REFERENCIAS BIBLIOGRÁFICAS
1. Gustafsson R. Systemic sclerosis. Aspects on pathogenetic mechanisms. Comprehensive summaries o Uppsala disertations from faculty of medicine (343). Uppsala, 1992.
2. Satos Nagaoka T, Hasegawa M, Tamatani T, Nakanishi T, Takigawa M. Serum levels of connective tissue growt a factor elevated in patients with systemic sclerosis: association with extent of skin sclerosis and severity of pulmonary fibrosis. J Rheumatol. 2000;27:149-154.
3. Zakrzewska Pniewska B, Jablonska S, Kowolska Oledeska E, Blaszczyk M, Hausmanowa Petruswicz I. Sympathetic skin responde in scleroderma, scleroderma overlap syndromes and inflamatory myopathies. Clin Rheumatol. 1999;18:473-80.
4. Winkelmann R. Pathogenesis and staging of scleroderma. Acta Derm Venereol. 1976;56:83-92.
5. Giordano M, Capelli L, Tirri G, Valti M. Vorschlag xueiner neven kla ssifizierung und nomenklatur der progredienten genera lisierten sklerodermie (PGS). Verh Dtschges Rheumathol. 1980;6:379-82.
6. Rodnan G, Jablonska S, Medsger T. Classification and nomenclature of progresive systemic sclerosis (scleroderma). Clin Rheum Dis. 1976;5:5-13.
7. Bernett A, Miller M, Little Jhonh G. A Sunsival study of patients with scleroderma diagnosed over 30 years (1953-1983): the valve of a simple cutaneous classification in the early stage of the disease. JR Heumatol. 1988;15:276-283.
8. Le Roy E, Black C, Fleismajer R, Jablowska S, Krieg T. Medsger. Sclerodenma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15:202-5.
9. Siegel RC. Esclerodermia. Clin Med NA. 1997;61:283-99.
10. Akagi A, Tajima S, Ishibzshi A, Yamaguchi N, Nagai Y. Expresion of type XVI collagen in human skin fibroblast: enhanced expresion in fibrotic skin desease. J Invest Dermatol. 1999;113:246-50.
11. Hanlon R, King S. Overview of the radiology of connective tissue disorders in children. Eur J Radiol. 2000;33:74-84.
12. Poormhing H, Lucas M, Medsger TA. Systemic sclerosis sine scleroderma: demographic, clinical and serologic features and survival in forty-eigth patients. Arthritis Rheum. 2000;43:444-51.
13. Anderson M, Cambell F, Hollis S, Moore T, Jayson M, Herrick A. Non -invasive assessment of digital vascular reactivity in patients with primary Raynaud's phenomenon and systemic sclerosis. Clin Exp Rheumatol. 1999;17:49-54.
14. Birbaum N, Rabin B, Bassion S. Histocompatibility antigents in progressive systemic sclerosis, (scleroderma). J Reumatol. 1977;4:425-8.
15. Mizutani HT, Nouchi N, Hamanaka H. Topical tocoretimate improved hypertrophic scar, skin in systemic sclerosis and morphea. J Dermathol. 1999;26:11-17.
16. Elst E, Van Sui J, Lekom Smit L, Oranje A. Treatment of linear scleroderma with oral 1,25 - dihidroxiviatmin (D3) (Calcitrol) in seven children. Pediate Dermatol. 1999;16:53-8.
17. Uziel Y, Feldman B, Yeung R, Caxer R. Metotrexate and corticosteroid therapy for pediatric localized scleroderma. J Pediatr. 2000;136:91-5.
18. Martini A, Maccario R, Ravelli A, Montagna D, Benedetti F, Bonetti F. Marked and sustained improvement two years after antologies stem cell transplatation in a girl with systemic sclerosis. Arthritis Rheum. 1999;42:807-11.
19. Tager R, Tikly M. Clinical and laboratory manifestations of systemic sclerosis (scleroderma) in black South africans. Rheumatology. 1999;38:397-400.
20. Vierra E, Cunnighan B. Morphea and localized scleroderma in children. Semin Cutan Med Surg. 1999;18:210-25.
Recibido.19 de junio de 2000
Aprobado:28 de junio de 2001
Rafael Pila Pérez. Hospital Provincial Manuel Ascunce Domenech. Camagüey, Cuba.


{kind=link}
{kind=link}
{kind=link}