










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Archivo Médico de Camagüey
versão On-line ISSN 1025-0255
AMC vol.10 no.2 Camagüey mar.-abr. 2006
CASOS CLÍNICOS
Síndrome de bardet-bieldet. Reporte de un caso
Bardet-Biedl's síndrome. A case report
Dr. Humberto Osorio Mariño; Dr. Francisco Ávila Riopedre; Dr.Carlos M. Sarduy; Dr.Carlos E. Arévalos
Hospital Provincial Clínico Docente Docente Manuel Ascunce Domenech. Camagüey, Cuba.
RESUMEN
Se presentan los hallazgos clínicos y de laboratorio de un paciente con síndrome de Bardet-Biedl. Los primeros síntomas aparecieron durante la adolescencia lo cual admite un modo de herencia autonómica recesiva. Al examen físico se constató polidactilia axial de manos y pies, retinosis pigmentaria, obesidad troncular, retraso mental e hipogonadismo, también se encontró diabetes mellitus no insulino dependiente, hipertensión arterial e insuficiencia renal crónica terminal. Se concluyó que esta enfermedad de inicio temprano en la vida puede causar ceguera en la adolescencia y fallo renal crónico en la edad adulta.
DeSC: SÍNDROME DE BARDET-BIEDL/etiología; RETINITIS PINGMENTOSA; POLIDACTILIA; RETRASO MENTAL; OBESIDAD, ADOLESCENCIA.
ABSTRACT
Clinical and laboratory findings of a patient with Bardet–Biedl`s syndrome are presented. The first symptoms appeared during adolescence which admits a mode of recessive autonomic heritage. In the physical examination axial polydactylia of hands and feet, retinistis pigmentosa, trunk obesity, mental retardation and hypogonadismo were found. Also, diabetes mellitus non – insuline dependent, arterial hypertension and terminal chronic renal failure were detected. This pathology of early occurrence in life may be cause of blindness in adolescence and chronic renal failure y the adultness.
DeSC: BARDET– BIEDL SYNDROME/etiology; RETINITIS PIGMENTOSA; POYDACTYLY; MENTAL RETARDATION; OBESITY; ADOLESCENCE.
INTRODUCCIÓN
John Z. Laurence y Robert Moon describen en 1866 a una familia londinense constituida por cuatro de ocho hermanos con retinosis pigmentaria, retraso mental, baja estatura para la edad e hipogonadismo en hombres; poco después algunos miembros de esta familia presentaron paraplejia espástica. Sin embargo, los primeros casos no fueron reportados hasta 1920 cuando George Bardet, 1 caracteriza la enfermedad con cuatro criterios: retinosis pigmentaria, polidactilia, obesidad e hipoplasia genital. En 1922 el endocrinólogo Arthur Biedl, 2 agrega el retraso mental. Solís Cohen y Weiss en 1925 acuñan el término Laurence Moon Bardet Biedl síndrome (LMBBS). La asociación con enfermedad renal fue descrita en detalle por Hurley, et al, 3 en 1975, esta constituye la sexta manifestación clínica del síndrome. Recientemente el síndrome ha vuelto a separarse en base a los hallazgos clínicos: Laurence Moon syndrome (LMS) se caracteriza por retinosis pigmentaria, retrazo mental, hipogonadismo y paraparesia espástica progresiva sin polidactilia. Bardet Biedl syndrome (BBS) se presenta con retiosis pigmentaria, obesidad troncular, polidactilia postaxial, deterioro cognitivo, hipogonadismo masculino, malformaciones genitourinarias femeninas complejas y disfunción renal, que es la mayor causa de morbimortalidad. La comunidad médica y científica ha adoptado actualmente esta división de la nomenclatura. Bealest et al, 4, 5 en 1999 identifican en un estudio retrospectivo de 109 pacientes, rasgos distintivos entre los enfermos con BBS, proponen para el diagnóstico clínico criterios primarios y secundarios según la frecuencia de aparición de estos.
El BBS se trasmite genéticamente a través de las familias por el modelo de herencia autonómico recesivo. En este tipo de herencia ambos padres llamados portadores tienen un gen del síndrome apareado con un gen normal, cada uno de sus hijos tiene entonces una probabilidad del 25 % de heredar los genes del síndrome. El pronóstico de la visión para los niños con BBS es malo. La ceguera nocturna usualmente se presenta entre los siete y ocho años de edad, y la aparición de la amaurosis bilateral a partir de los 15 años. El peso al nacer es normal, la ganancia de peso comienza en el primer año y se mantiene para toda la vida en la mayoría de los enfermos. Casi la totalidad de los enfermos tienen dificultad en el aprendizaje, pero sólo la minoría tiene severos trastornos cognitivos.
El diagnóstico de BBS es clínico. 5 Se conocen cinco genes asociados con el síndrome: BBS1, BBS2, BBS4, MKKS (BBS6) y BBS7. Dos locus adicionales BBS3 y BBS5 tienen vínculos también.
Hay que diferenciar esta enfermedad de otros dos síndromes: McKusick Kaufman syndrome (MKKS): se caracteriza por la tríada de hidrometrocolpus, polidactilia postaxial y cardiopatía congénita, su causa es por mutación del gen MKKS. 6 El segundo diagnóstico diferencial lo constituye el Alström syndrome: retinosis pigmentaria, obesidad, sordera sensorial progresiva, cardiomiopatía dilatada, diabetes mellitus tipo dos y desarrollo motor tardío, a diferencia del BBS no hay deterioro cognitivo ni polidactilia tratándose de una mutación en el gen ALMS y se trasmite con carácter autosómico recesivo. 7
PRESENTACIÓN DEL CASO
Paciente femenina, blanca, de 65 años de edad. Con un peso de 46 Kg y talla de 126 cm, con antecedentes de retraso mental ligero, polidactilia en los cuatro miembros, pérdida total de la visión desde los doce años de edad, anemia e hipertensión arterial diagnosticadas hace tres años. En el mes de noviembre de 2003 acudió a nuestro centro remitida desde el hospital municipal de Florida, refirió tener astenia, anorexia y náuseas. Al examen físico constató un soplo sistólico eyectivo aórtico 2/6, hipertensión arterial y cifras de creatinina elevada con disminución del gasto urinario.
APF: Hermano vivo que presenta amaurosis bilateral, polidactilia y retraso mental.
Padre: fallecido con antecedentes de hipertensión arterial.
Madre: fallecida con antecedentes de hipertensión arterial.
Exámenes complementarios:
Hb: 67g/l.
Creatinina: 601, 2 mcmol/l.
Glucemia: 12, 1 mmol/l.
VSG: 60 mm/h.
Ácido úrico: 361 mmo/l
Proteínas totales: 60 g/l
Lipidograma: bpre b: 0, 60 uds.
TG: 3, 2 mmol/l.
Colesterol: 7, 4 mmol/l
Minicultivo: negativo.
Proteinuria de 24 h: Trazas ) no dosificables).
Conteo de Addis: Proteínas: ligeras trazas.
Leucocitos: 48100.
Hematíes: 1100.
Cilindros: 0.
Rx de tórax: Aumento del índice cardiotorácico a expensa del ventrículo izquierdo.
Hilios pulmonares congestivos.
Fondoscopia:
OD: espículas satélites a los vasos, osteoblastos y palidez pupilar típico de retinosis pigmentaria.
OI: opacidad total del cristalino por catarata.
ERG: extinguido en OD.
USD: abdominal y ginecológico: hígado homogéneo que no rebasa el reborde costal derecho, vesícula estimulada sin litiasis, páncreas y bazo normales, ambos riñones de pequeño tamaño y ecogénicos. Aorta abdominal de calibre normal. Útero hipoplásico (50x19x32 mm). Anejos de muy pequeño tamaño.
Test psicométrico: retraso mental ligero-moderado.
DISCUSIÓN
La paciente presentó manifestaciones clínicas del BBS, con características distintivas dadas por criterios primarios y secundarios. 4, 5, 8
Se encontraron diversos criterios primarios para el diagnóstico, entre ellos: la retinosis pigmentaria que presenta las distrofias de conos y bastones, es el signo más temprano de disfunción de la retina, a menudo no aparece hasta los siete u ocho años de edad en forma de hemeralopia y llevan a un deterioro visual severo en la madurez joven, otros signos clínicos extrarretinianos incluyen: nistagmo, estrabismo, miopía, catarata y glaucoma. La polidactilia es común pero su presencia no es invariable; suele aparecer entre el 58-69 % de los enfermos 6, la forma más común es la postaxial hexadactilia presente en la paciente. Puede hallarse braquidactilia así como sindactilia (más común entre el segundo y tercer dedo). Con cierta frecuencia los dedos de las manos y los pies son cortos y gruesos con formación de piel extra en forma de anillo en la base de los dedos de los cuatro miembros (Figura 1).
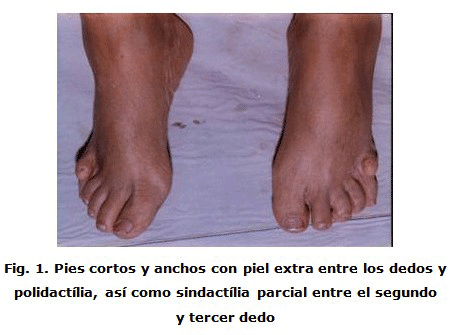
La obesidad puede estar presente desde la niñez, suele ser más difusa en adolescentes pero se va haciendo troncular y proximal de miembros en la edad adulta (Figura 2).

Los trastornos del aprendizaje experimentan dificultad en su desarrollo cognitivo y el retraso mental suele ser ligero-moderado. Según el hipogonadismo masculino o anormalidades genitales en mujeres se precisan cuando alcanzan la pubertad. Los varones con BBS, tienen genitales pequeños (pene y testículos). Las mujeres pueden presentar hipoplasia de trompas de Falopio, útero y ovarios, atresia total o parcial de vejiga, fístula vesicovaginal, himen imperforado, ausencia de orificio uretral 9, pueden experimentar ciclos menstruales irregulares. Las anormalidades estructurales y funcionales renales incluyen el doble sistema pielocalicial, divertículo de vejiga, hipoplasia renal, reflujo vesicouretral y fallo renal crónico. 10
Dentro de los criterios diagnósticos secundarios se encuentran el inicio tardío del habla en la niñez y dificultad en la fonación, estrabismo y astigmatismo, desarrollo motor tardío, ataxia e incoordinación, cardiopatía congénita y fibrosis hepática. 11
Esta paciente se corresponde con BBS, como lo describe la literatura y lo confirman los criterios diagnósticos primarios presentes en la enferma. El BBS constituye una afección poco frecuente en este medio, mientras que la insuficiencia renal crónica es relativamente común y solo fue planteado cuando apareció el fracaso renal; complicación que con mayor frecuencia lleva a la muerte a estos individuos.
REFERENCIAS BIBLIOGRÁFICAS
1. Bardet G. Sur un syndrome d'obesite congenitale avec polydactylie et retime (contribution a I'etude des formes clinique de l'obesite hypophysaire.) Paris: Sobornne; 1920.
2. Biedl A. Ein Geschwisterpaar mit adiposo-genitaler dystrophone. Dtsch med wschr. 1922;48:1630.
3. Hurley RM, Borruat FX, Apathy PP. Patterns of rod and cone dysfuntion in Bardet-Biedl syndrome. Am J. Ophthalmol. 1990;109:676-88.
4. Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved of Bardet-Biedl syndrome: results of a population survey. J Med Hum Genet. 1999;36:437-46.
5. Beales PL, Katsanis N, Lewis RA, Ansley SJ, Elcioglu N. Raza J, et al. Genetic and mutational analyses of a large multiethnic Bardet-Biedl cohort reveal a minor involvement of BBSG and delineate the critical intervals of other loci. Am J Hum Genet. 2001;68:606-16.
6. Beales PL, Badano JL, Roos AJ, Ansley SJ, Hoskins BE, Kirsten B, et al: Genetic interaction of BBS-1 mutations with alleles at other BBS loci can results in non-Mendelian Bardet-Biedl syndrome. Am J Hum Genet. 2003; 72: 1187-99.
7. Mykytyn K, Nishimora DY, Searby CC, Beck G, Bugge K, Haines HL, et al: Evaluation of complex inheritance involving the most commun Bardet-Biedl syndrome locus BBS1. Am J Hum Genet. 2003;72:429-37.
8. Hoskins BE, Thorn A, Scambler PJ, Beales PL. Evaluation of multiplex capillary heteroduplex analysis; a rapid and sensitive mutation screening technique. Hum Mutat. 2003; 22: 151-57.
9-. Dar P, Sachs GS, Carter SM, Ferreira JC, Nitowky HM, Groos SJ. Prenatal diagnosis of Bardet-Biedl syndrome by targeted second trimester sonography. Ultrasound Obstet Gynecol. 2001;17:354-56.
10. Badano JL, Ansley SJ, Leitch CC, Lupski JR, Katsanis N. Identification of a novel Bardet- Biedl syndrome protein BBS7, that shares structural features with BBS1 and BBS2. Am J Hum Genet 2003; 72: 650-58.
11. Katsanis N, Ansley SJ, Badano JL, Eichers ER, Lewis RA, Hoskins BE, et al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293:2256-59.
Recibido: 20 de octubre de 2004
Aprobado: 20 de diciembre de 2004
Dr. Humberto Osorio Mariño. Especialista de I Grado en Nefrología. Hospital Provincial Clínico Docente Docente Manuel Ascunse Domenech. Camagüey, Cuba.
