










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Archivo Médico de Camagüey
versión On-line ISSN 1025-0255
AMC v.14 n.5 Camagüey sep.-oct. 2010
CASOS CLÍNICOS
Síndrome de Sturge-Weber: presentación de un caso
Dr. Rafael Pila PérezI; Dr. Mario E. Conde RiveraII; Dr. Rafael Pila PélaezIII; Dr. Victor Holguín PrietoIV; Dr. Etelivar Torres VargasV I Residente de I año en Medicina Interna. Hospital Universitario Manuel Ascunce Domenech. Camagüey, Cuba. II Residente de III año en Medicina Interna. III Especialista de II Grado en Medicina Interna. Profesor Titular. IV Especialista de II Grado en Medicina Interna. Profesor Auxiliar. V Residente de III año en Medicina Interna.
RESUMEN
Fundamento: el síndrome de Sturge-weber es una enfermedad muy rara y cuyo signo cardinal es la " Mancha en vino de Oporto". Objetivo: presentar un caso inusual de un paciente de ochenta años de edad con síndrome de Sturge-Weber, se hospitalizó en múltiples ocasiones por presentar severas alteraciones oftalmológicas y neurológicas. El caso se clasificó como un síndrome de Sturge-Weber tipo I. Método: se presenta un paciente, sexo masculino, ochenta años y antecedentes de "Mancha en vino de Oporto" desde su nacimiento, con crisis epiléptica, cefalea casi constantes, crisis de isquemia vascular transitoria en múltiples ocasiones e ingresado por accidente vascular encefálico isquémico en dos oportunidades como resultado de la misma, una hemiplejía izquierda residual. El diagnóstico se realizó por la clínica, tomografía axial computarizada, electroencefalograma y características del LCR. Llamó la atención la no presentación de deterioro mental grave. Conclusiones: ante la presencia en la niñez y la juventud de una " Mancha en vino de Oporto" en la región del nervio trigémino, obliga a un estudio exhaustivo del paciente para lograr mejorías oftalmológicas, neurológicas y cutáneas con la terapéutica actual, sino se logran los resultados deseados, pues se debe utilizar la cirugía como medio para evitar el deterioro mental y oftalmológico, el cual puede ocasionar pérdida de la visión. DeCS: SINDROME DE STURGE-WEBER/diagnóstico; ANCIANO; MANCHA VINO DE OPORTO; ESTUDIOS DE CASOS
ABSTRACT
Background: Sturge-Weber syndrome is a very rare disease whose cardinal sign´s is the “port-wine stain". Objective: to present an unusual case of an eighty years old patient with a Sturge-Weber syndrome who has been admitted in several occasions for presenting severe ophthalmological and neurological alterations that warns to deepen in the study of these patients. The case was classified as a type I Sturge-Weber syndrome. Method: an eighty year-old male with “port-wine stain" antecedents from his birth, with epileptic seizure, almost constant cephalea, crisis of transient vascular ischemia in multiple occasions and admitted by ischemic encephalic vascular accident in two opportunities as a result of this, a residual left hemiplegia. Diagnosis is carried out by the clinic, computerized axial tomography, electroencephalogram and LCR characteristic. Attracting the attention he did not present a serious mental impairment. Results: important finding in this syndrome is the presence of “port-wine stain” with convulsions, glaucoma, cephalea and mental impairment. At present neuroimaging study is fundamental. Conclusions: under the presence of a “port-wine stain" in the region of the trigeminal nerve in childhood and youth forces to the patient's exhaustive study to achieve ophthalmological, neurological and cutaneous improvements with the current therapy and if wanted results are not achieved, to use surgery as mean of avoiding ophthalmologic and mental impairment that may reach to the loss of vision like it happened to our patient.
DeCS: STURGE-WEBER SYNDROME/diagnosis; AGED; PORT-WINE STAIN; CASE STUDIES
INTRODUCCIÓN
El Sindrome de Sturge-Weber (SSW) también llamado angiomatosis encefalotrigeminal, es un desorden neurocutáneo con angiomas que afectan las leptomeninges y la piel de la cara, típicamente las zonas oftálmicas y maxilar que son distribuidos por el nervio trígemino. El angioma cutáneo es conocido como "Mancha en vino de Oporto". 1, 2 Se puede presentar también en angiomas extracraneales y de tejidos blandos. 1 Otras malformaciones del sistema nervioso central (SNC) se asocian a esta enfermedad y otros síndromes neurocutáneos se incluyen en su diagnóstico diferencial. 2
El SSW es conocido como completo cuando el SNC y los angiomas faciales están presentes y son incompletos cuando sólo uno está presente; la escala de Roach se utiliza para esta clasificación: 3
- Tipo I. Angioma facial y de leptomeninges están presentes; puede tener glaucoma.
- Tipo II. Angioma facial solamente, no SNC afectado; puede tener glaucoma.
- Tipo III. Angioma de leptomeninges solamente; no glaucoma.
El SSW es causado por vasos sanguíneos embrionarios residuales y sus efectos secundarios alrededor de los tejidos cerebrales. Se desarrolla un plexo vascular alrededor de la porción cefálica del tubo neural debajo del ectodermo destinada a convertirse en la piel de la región facial, normalmente este plexo vascular se forma a las seis semanas y vuelve al estado anterior a las nueve semanas de la gestación. Como enfermedad de esta regresión se presenta un tejido vascular residual que forma los angiomas de las leptomeninges, cara y ojo ipsilateral. La disfunción neurológica resulta del efecto secundario del tejido cerebral rodeado por esta alteración que incluye hipoxia, isquemia, oclusión venosa, trombosis, infarto o fenómeno vasomotor. 1, 4, 5 Norman y Schorne6 piensan que la anormalidad en el flujo sanguíneo en la angiomatosis de las leptomeninges causan aumento de la permeabilidad capilar, estasis y anoxia, García, 7 Gómez y Besin, 8 reportan que la oclusión venosa pueden causar los eventos neurológicos iniciales tales como convulsiones y hemiparesias o ambos y pueden dar inicio a este proceso un "fenómeno de robo vascular", puede desarrollarse alrededor del angioma dando como resultado isquemia cortical, por tanto las convulsiones recurrentes, estados epilépticos, convulsiones intratables y eventos vasculares recurrentes pueden agravar este estado con incremento de la isquemia cortical, dando por resultado calcificaciones, gliosis y atrofias con cambios en la presencia de convulsiones y deterioro neurológico. 1-3 La progresión de la enfermedad y el deterioro neurológico se puede presentar en el SSW y ha sido documentada por varios autores, 7-9 sin embargo, el control de las convulsiones y el deterioro neurológico se han controlado con la aspirinoterapia y el tratamiento quirúrgico. 9
Las manifestaciones oculares más importantes son el buftalmos y glaucoma que son secundarios al aumento de la presión intraocular con una obstrucción mecánica del ángulo del ojo, aumento de la presión episcleral o secreción elevada de los fluidos acuosos. 4 Las causas del SSW no están claras, pero se han reportado evidencias de mosaisismos somáticos en muchos pacientes, sobre todo inversión del brazo 4q del cromosoma y trisomía 10. 1, 10
El objetivo de este trabajo es presentar el caso de un paciente de 80 años con el cuadro clínico de esta enfermedad desde su niñez y que no fue diagnosticado y tratado correctamente hasta este momento.
CASO CLÍNICO
Se presenta un paciente de sexo masculino, 80 años, obrero agrícola hasta la jubilación, con múltiples ingresos en su niñez, juventud y adultez en el antiguo hospital de La colonia Española y después en otros hospitales de esta ciudad y de ciudad de La Habana, con antecedentes de angioma facial izquierdo desde su niñez, epilepsia, accidente vascular isquémico en dos oportunidades, crisis de isquemia vascular transitoria en múltiples ocasiones, cefalea constantes, de padecer de diabetes mellitus desde los 50 años, glaucoma desde los 25 años, se ingresa en el Hospital Universitario Manuel Ascunce Domenech por accidente vascular cerebral isquémico (ACVI) sin presentar en esta ocasión convulsiones.
Examen Físico
Paciente consciente orientado, que coopera el interrogatorio, disártrico, con buen estado general que presenta hemangioma completo de la hemicara y hemicráneo izquierdo, y porción del hemicráneo derecho y discreta parte de esta hemicara con pérdida total de la visión del ojo izquierdo. (Figura 1)
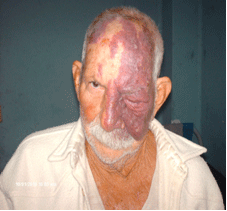
Fig 1. Mancha facial en vino de Oporto, afectación del ojo izquierdo y los nervios inervados por el trigémino
No se encuentran angiomas en ningún otro sitio del cuerpo. El examen físico es totalmente normal en aparatos y sistemas excepto el sistema nervioso donde encontramos un paciente disartrico, con dificultad de la marcha por accidente vascular encefálico isquémico antiguo y con hemiplejía izquierda espástica a predominio crural, hemangioma facial y craneal izquierdo con daño del nervio trígemino y sus ramas oftálmicas y maxilares y perdida total de la visión de dicho ojo; no otras alteraciones. Tacto rectal: hiperplasia benigna de la próstata grado II. Fondo de ojo: perdida de visión del ojo izquierdo. Glaucoma del ojo derecho y microhemorragia en el cuadrante nasal superior de dicho ojo.
Estudio analítico:
Exámenes de la laboratorio hemoquímica, enzimas hepáticas, pancreáticas función renal, iones, VDRL, VIH: negativos. Coagulación, sangramiento, estudio de inmunidad, cultivos de sangre y orina dentro de la normalidad. Radiografía de tórax, ECG, y ultrasonido abdominal y de próstata dentro de la normalidad, la última aprecia un aumento discreto de la glándula prostática. El estudio del líquido cefalorraquídeo solo mostró aumento moderado de proteínas. TAC de cráneo. (Figura 2)

Fig 2. Existe calcificación parietal posterior derecho que describe un trayecto vascular en relación con malformación arterio-venosa. Atrofia severa del hemisferio cerebral derecho. Sistema ventricular normal; no desplazamiento de estructuras cerebrales
Existe calcificación parietal posterior derecha que descubre un trayecto vascular en relación con malformación arteriovenosa. Atrofia severa el hemisferio cerebral derecho. Sistema ventricular normal sin desplazamiento de las estructuras cerebrales. Se intenta realizar TAC contrastada y el paciente se niega. Electroencefalograma: Atenuación marcada del voltaje en región cerebral izquierda y actividad polimórfica delta, propia de epilepsia crónica.
En este paciente se emplearon para su tratamiento anticonvulsivantes del tipo de carbamazepina, antiagregante plaquetario del tipo de dipiridamol y aspirina, así como gotas oftálmicas de B- antagonistas y fisioterapia especialmente en el hemicuerpo izquierdo. El mismo fue egresado mejorado a los 25 días de su ingreso, siendo atendido por la consulta de glaucoma en el departamento de Oftalmología y por Medicina Interna.
DISCUSIÓN
No se reporta diferencia racial; ambos sexos pueden ser afectados y el típico paciente con angioma facial se presenta desde el nacimiento. 1Como ocurrió en este enfermo pero no todos los niños con angiomas faciales tienen un SSW, 8 y se presenta en casi 5x100.000 nacidos vivos.2
El angioma facial es una lesión macular congénita que es de crecimiento progresivo, puede ser rosada en sus inicios, pero puede progresar a una forma de color oscura y nodular, esta puede ser única o asociada con lesiones de los vasos coroideos de los ojos o de las leptomeninges del cerebro o localizarse en otras aéreas del cuerpo, 2, 3 en nuestro caso fue única y localizada en cara y cráneo del lado izquierdo y parte del lado derecho. La incidencia del SSW con angioma facial se ha reportado entre el 8-33% de los casos solamente. 1 Enjoirías, et al,11 en un estudio retrospectivo de 106 enfermos con angiomas faciales encontró 12 que tenían SSW y cuatro con glaucoma y sin lesión de leptomeninges. Bioxeda, et al 12 en un estudio de 121 pacientes encontró que el 86% eran unilaterales y bilaterales en el 14% y la epilepsia y el glaucoma estuvieron presentes en el 17% y 14% respectivamente. En el mayor estudio de esta enfermedad realizado por Tallman, et al 13 en 310 pacientes con angioma facial el 85% fue unilateral y el 15% bilateral. Nuestra paciente lo presenta de forma bilateral, aunque no es muy marcada en la hemicara derecha, pero si en el hemicráneo derecho.
Estudios realizados por Sujansky y Conradi 14 en 52 adultos y que fueron identificados por la Fundación de Sturge-Weber encontraron convulsiones en el 83%, glaucoma en el 60% y déficit neurológico en el 65% incluyendo accidentes vasculares, parálisis, disartrias, espasticidad y debilidad como en nuestro paciente. La edad de comienzo en estos pacientes de las convulsiones estuvo en el rango del nacimiento a los 23 años, con una media de seis meses, 15 este paciente lo señala desde los diez años más o menos. Se señalaron los controles de las convulsiones con diferentes medicamentos en el 27%, controles a mediano plazo o de forma irregular en el 49% y que y que no mejoraron en el 24%, 5-9 este paciente ha tenido en realidad un control no total pues ha sido ingresado en varias ocasiones por esta manifestación. El dolor de cabeza ocurre por lo regular en el 62% de los enfermos; la frecuencia puede ser diaria, de una a cuatro veces a la semana y una o dos veces al mes, 9 nuestro paciente la presentó uno o dos veces por semana. El dolor de la cabeza se asocia con el aumento de la descoloración del angioma facial y puede acompañarse de auras, nauseas, vómitos, disartria, mareos y sensación de un pulso facial, 1, 9 nuestro paciente señaló siempre disartria y mareos.
El comienzo del glaucoma ocurre entre 0-41 años, con una media de cinco años, 7 este enfermo lo presentó a los 40 años y abandonó el tratamiento del oftalmólogo, por lo que presentó perdida de la visión del ojo izquierdo, el glaucoma se reporta entre el 30-71%. 1, 2, 15 Es llamativo que en esta enfermedad se producen alteraciones mentales tales como retraso mental en el 50-75%, 1 este paciente no presentó estas alteraciones y mantuvo status laboral hasta su retiro definitivo.
El diagnostico diferencial debe realizarse con los sindromes de Klippel-Trenaunay-Weber; Rendu-Osler-Weber; Von Hippel- Lindau; Wyburn-Mason; Shapiro- Shulman; Divry- Van Boagaert; Bannayan- Zonna y Sindrome de Cobb. 1, 12, 13
Los exámenes de laboratorio son por lo regular normales como ocurrió en este caso, pero el estudio del LCR puede presentar proteínas elevadas, presumiblemente por microhemorragia las cuales son comunes en el SSW, no así las hemorragias importantes, 1 en nuestro caso el estudio del LCR, mostró elevación de proteínas pero no sangramiento. La radiografía de cráneo puede mostrar la clásica " Tram-line" o de "Tram- Track" de las calcificaciones que se consideraban patognomónicas del SSW, antes de la era moderna de la neuroimagen, 16 pero hoy la tomografía axial, muestra estas calcificaciones, atrofia cortical y drenaje venoso anormal. En un estudio realizado por Terdjaman, et al, 17 en 14 pacientes, 12 presentaron calcificaciones y diez atrofia cortical. Sin embargo, la MRI con gadolinio permite el diagnóstico en el recién nacido con angiomatosis facial, 16 no muestra las calcificaciones y sí los angiomas de las leptomeninges. El electroencefalograma se utiliza para la evaluación de las convulsiones, sobre todo para formas localizadas, refractarias y cuando en la epilepsia es considerada la cirugía. 18 Brenner y Shanbrough, 19 reportan el antecedente de la reducción unilateral de la amplitud como el hallazgo más frecuente, sin embargo, Sassower, et al18 señalan una marcada atenuación del voltaje en la región del angioma, y actividad delta polimórfica en 13 de sus 14 pacientes y como se encontró en nuestro enfermo.
El tratamiento de forma general incluye los anticonvulsivantes para el control de la epilepsia, tratamiento sintomático y profiláctico para el dolor de cabeza y glaucoma para reducir la presión intraocular; y terapia con láser para el angioma facial. 1, 14, 17 Este paciente tiene para su terapéutica anticonvulsivante: fenobarbital, fenitoina, carbamazepina, clonazepan, gabapentina, etc, en estos momentos se emplea de ellas la carbamazepina. En el caso del glaucoma lo fundamental es reducir la presión intraocular por lo que se emplea gotas oftálmicas B-antagonistas (Timolol). El dolor de cabeza uno de los signos más frecuentes en estos enfermos a veces requieren de varios medicamentos para su alivio tales como ibuprofeno, aspirina, amitriptilina, derivados del cornezuelo de centeno, etc, 1, 9, 13 pero Kossoff, et al 20 señalan que a veces no existe un medicamento adecuado para estos enfermos, por ello para prevenir el dolor de cabeza, y las crisis de accidente vascular periférico se emplea la aspirina. Thomas –Sohl, et al 21 recomiendan una dosis de 3-5 mg/kg/día para las crisis de ataques transitorios de isquemia, y para la cefalea.
Las "Manchas en vino de Oporto" debe ser evaluadas en la primera semana de la vida y diferenciarla de los hemangiomas; las manchas se tratan con laserterapia tan pronto sea posible, pues no sólo es el mejor tratamiento para la lesión sino el logro de beneficios psicológicos importantes.2, 3, 22 Cuando es difícil controlar las convulsiones con medicamentos, la extirpación quirúrgica temprana de la porción afectada del cerebro puede mejorar su control y evitar el deterioro mental, 1, 7, 8, 15 al igual que ocurre con el glaucoma u otras alteraciones asociadas. 1, 6,9 Los procederes quirúrgicos incluyen resección cortical focal, hemiesferectomia,corposcallosotectomía y estimulación del nervio vago. 23
REFERENCIAS BIBLIOGRÁFICAS
1.Parsa CF. Sturge Weber syndrome: A unififield patholophysiologic mechanism. Curr Treat Options Neurols 2008;10:47-54.
2.Sharan S, Swamy B, Taranath D. Port Winc Vascular malformations and glaucoma risk in Sturge-Weber syndrome. JAAPOS 2009;13(4):374-8.
3.Roach ES. Neurocutaneuos syndromes. Pediatric Clin North AM 1992;39(4):591-620.
4.Govori V,Gjikolli B,Ajvazih H,Morina N.Management of patient with Sturge-Weber syndrome:A case report.Cases J 2009;2:93-95.
5.Comi AM. Pathophisiology of Sturge-Weber syndrome. J Child Neurol 2005; 20(8):509-16.
6.Norman M, Schoene W. The ultrastructure of Sturge- Weber disease. Acta Neurophatol 1977; 37(3):199-205.
7.García J, Roach E, Mc Jean W. Recurrent thrombotic deterioration in the Sturge- Weber syndrome. Childs Brain 1981; 8(6):427-33.
8.Gómez M, Benin E. Sturge-Weber syndrome. In Butterworths B. Neurocutaneous diseases. New York: A Practical Approch; 1987.p. 356-367.
9.Roach E, Bodensteiner JB. Neurologic manifestations of Sturge-Weber syndrome. In: Freedom M. Sturge-Weber syndrome. New Jersey: Sturge-Weber Foundation; 1999.p. 27-38.
10.Huq A, Chugani D, Hukku B. Evidence of somatic mosarcism in Sturge-Weber syndrome. Neurology 2005; 59(5):780-2.
11.Enjoiras O, Riche M, Merland J. Facial port-wine stains and Sturge-Weber syndrome. Pedatrics 1985; 76(1):48-51.
12.Bioxeda P, De Misa R, Arrazola J. Facial angioma and the Sturge-Weber sydrome: a study of 121 cases. Med Clin (Barc) 2006; 111(1):1-4.
13. Tallman B, Tan O Morelli J. Location of port-wine stains and the likelihood of ophthalmic and/or central nervous system complications. Pediatrics 1997; 93(3):323-7.
14.Sujansky E, Conradi S. Outcome of Sturge-Weber syndrome in 52 adults. Am J Medgenet 1999; 61(1):35-45.
15.Jung A, Raman A, Rowland Hill C. Acute hemiparesis in Sturge-Weber syndrome. Neurol 2009; 9(3):169-71.
16.Udani V, Pujars H, Munot P, Maheshwaris H, Mehta N. Natural history and magnetic resonance imaging follow up in 9 Sturge-Weber syndrome patients and clinical correlation.J Child Neurol 2007;32(4):479-83.
17.Terdman P, Alcardi J, Sainte-Rose C. Neuroradiological findings in Sturge-Weber syndrome (SWS) and isolated pial angiomatosis. Neuropediatrics 2005; 35(3):115-20.
18.Sassower K, Duchowhy M, Jayakar P. EEG evaluation of children with Sturge-Weber syndrome and Epilepsy. J Epylepsy 2008; 23:285-289.
19.Brenner RP, Sharbrough PW. Electroencephalographic evaluation in Sturge-Weber syndrome. Neurology 1976; 26(7):629-32.
20.Kossoff EH, Balasta M, Haffield L, Lehmann CU, Comi AM. Self-Reported treatment patterns in patient´s wifh Sturge-Weber syndrome and migraines. J Child Neurol 2007; 22(6):720-6.
21.Thomas Sohl K, Vaslow D, Maria BL. Sturge-Weber syndrome: A review. Pediatric Neurol 2006; 30(5):303-10.
22.Audren F, Abitbal O, Dureau P. Non-penetranting deep sclerectomy for glaucoma associated with Sturge-Weber syndrome. Acta Ophthalmol Scand 2006; 84(5):656-60.
23.Kossoff EH, Buck C, Freeman JM. Outcome of 32 hemispherectomies for Sturge-Weber syndrome worldwide. Neurology 2005; 59(11): 1735-8.
Recibido: 8 de abril de 2010 Aprobado: 2 de julio de 2010
Dr. Rafael Pila Pérez. Residente de I año en Medicina Interna. Hospital Universitario Manuel Ascunce Domenech. Camagüey, Cuba
