










My SciELO
 Custom services
Custom servicesServices on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Archivo Médico de Camagüey
On-line version ISSN 1025-0255
AMC vol.15 no.3 Camagüey May-.June 2011
CASO CLÍNICO
Síndrome de Stevens-Johnson: presentación de un caso
Stevens-Johnson syndrome: a case presentation
Dr. Ener de Jesús Fernández BrizuelaI; Felicia Morales DíazII
I Especialista de I Grado en Pediatría. Máter en Atención Integral al niño. Profesor instructor. Policlínico Universitario Teniente Tomás Rojas. Carlos Manuel de Céspedes.Céspedes, Camagüey, Cuba. fbener@finlay.cmw.sld.cu II Especialista de I Grado en MGI. Master en Longevidad satisfactoria. Profesor Instructor Policlínico Universitario Concepción Agramonte Bossa. Florida, Camagüey, Cuba.
RESUMEN
Fundamento: el síndrome de Stevens-Johnson, también llamado eritema multiforme mayor, se caracteriza por una serie de lesiones cutáneas de diversa morfología, es una grave lesión en la cual están afectadas al menos dos membranas mucosas y la piel. La patogenia de esta enfermedad es desconocida aunque generalmente se considera que es provocada por una reacción de hipersensibilidad a drogas, infecciones y exposición a sustancias tóxicas. Caso clínico: esta afección fue diagnosticada en un niño africano de nueve años de edad. El niño desarrolló una severa y florida forma de ulceración mucocutánea, manifestaciones orales y oculares serias, marcados trastornos sistémicos y deshidratación. Se realizó una amplia revisión bibliográfica sobre el tema donde fueron verificadas las manifestaciones clínicas, evolución y el tratamiento de esta enfermedad.
DeSC: SINDROME DE STEVENS-JOHNSON/diagnóstico; SINDROME DE STEVENS-JOHNSON/complicaciones; INFORMES DE CASOS; NIÑO, ERITEMA MULTIFORME
ABSTRACT
Background: Stevens-Johnson syndrome also called erythema multiforme major, it is characterized by a series of cutaneous lesions with different morphology, and it is a serious lesion in which two mucous membranes and skin are affected at least. The pathogeny of this disease is ignored although it is generally considered that it is caused by a drug hypersensitivity reaction, infections and exposure to toxic substances. Clinical case: this affection was diagnosed in a nine year-old African boy. The boy developed a severe and florid form of mucocutaneous ulceration, serious oral and ocular manifestations, marked systemic disorders and dehydration. A wide bibliographical review on the topic was carried out where clinical manifestations, evolution and treatment of this disease were verified.
DeSC: STEVENS-JOHNSON SYNDROME /diagnosis; STEVENS-JOHNSON SYNDROME/complications; CASE REPORTS; CHILD, ERYTHEMA MULTIFORME
INTRODUCCIÓN
El síndrome de Stevens-Johnson, también llamado eritema multiforme mayor, fue descrito por vez primera en 1992 y caracterizado por una serie de lesiones de piel de diversa morfología, en la cual están severamente dañadas al menos dos membranas mucosas y la piel. La incidencia es probablemente de uno a tres casos por cada millón de habitantes al año.1
La patogenia de esta enfermedad es desconocida, aunque en sentido general se plantea una reacción de hipersensibilidad relacionada con drogas, infecciones y exposición a sustancias tóxicas. Los agentes tóxicos que han sido concluyentemente demostrados han sido el virus del herpes simplex y el mycoplasma pneumoniae. No obstante en el 20 % de los casos no se puede probar ninguna de sus causas.2-4 Otras causas que se relacionen con esta enfermedad son: 2-5
Infecciones: virus del herpes simplex, mycoplasma pneumoniae, mycobacterium tuberculosis, streptococos del grupo A, virus de la hepatitis B, virus Epstein Barr, francisella tularensis, yersinia, histoplasma, enterovirus y VIH.
Drogas: penicilinas, sulfamidas, tetraciclinas, isoniazida, fenitoína, fenobarbital, carbamazepina, ácido acetil salicílico, captopril, fenilbutasona.
Neoplasias: leucemia y linfomas.
Otros: radioterapia, embarazo y luz solar.
La relación de ocurrencia entre varones y hembras es de dos a uno. La reacción farmacológica ocurre de una a tres semanas después de la exposición.5
La recuperación completa requiere de cuatro a seis semanas y la recuperación de las lesiones de piel cursa con híper o hipopigmentación pero sin dejar cicatrices.5-6
Las complicaciones más frecuentes suelen ser la estenosis esofágica y la pérdida de visión por úlceras corneales.7
Aproximadamente de un 25 a un 50% de los casos tienen solo afectada la mucosa oral, con especial predilección por el borde rojo de los labios y la mucosa oral y raramente afecta las encías. La mayoría de los pacientes sufre un solo episodio de eritema multiforme mayor. La recurrencia se relaciona con la exposición a drogas y a la infección por el virus del Herpes simplex.8
Un número importante de casos requieren ingreso en unidades de cuidados intensivos con fluidos endovenosos, soporte nutricional, ropas ligeras, camas especiales, analgesia y cateterismo vesical si necesario. Los antibióticos son muy importantes en las infecciones bacterianas secundarias que en ocasiones pueden provocar la muerte. La mortalidad oscila entre el 5 y el 15 % de los casos.8
CASO CLÍNICO
Se presenta el caso del paciente C. M. de nueve años de edad, sexo masculino y color de la piel: negra. El paciente se atendió en los servicios de pediatría del Hospital infantil de Harare de la provincia distrital de Harare en Zimbabue en los meses de noviembre y diciembre de 2007. Acudió al Hospital por cefalea intensa de tres días de evolución y rash generalizado en la piel de dos días de evolución lo cual constituyó el motivo de ingreso, y cuya historia de la enfermedad actual se describió como paciente con antecedentes personales de salud hasta tres días atrás donde comenzó con cefalea frontal y un hermano de 11 años le ofreció una pastilla del bolso de su mamá sin precisar el nombre, y al día siguiente se levanta con ampollas generalizadas y no pruriginosas, asociadas con fiebre, escalofríos y confusión mental ocasional. El padre refierió que el niño presentó dificultad para orinar pero frecuencia normal, poco apetito y vómitos después de ingerir alimentos. No diarreas, no tos, no convulsiones. Antecedentes perinatales de parto a término y peso de 3600g al nacer con un puntaje de Apgar normal al minuto y a los cinco minutos del nacimiento.
Antecedentes patológicos personales: inmunizaciones actualizadas. No historia de alergia medicamentosa. Es el cuarto hijo de un matrimonio con cuatro varones y una hembra.
No historia familiar de VIH, asma, alergia o tuberculosis. Resto de los hermanos normales.
Al examen físico: paciente críticamente enfermo. Febril con 38,2º Celsius y escalofríos. Peso 20Kg. Piel: abundantes flictenas rotas en su mayoría, con predominio en la cara y afectación de la mucosa oral, conjuntival y los genitales. Las ampollas no estaban sépticas. Signos evidentes de deshidratación moderada con mucosas secas. (Figuras 1, 2 y 3).



Sistema respiratorio: respiración espontánea, no dificultad respiratoria, murmullo vesicular normal, no estertores, frecuencia respiratoria: 22 respiraciones por minuto. Sistema cardiovascular: ruidos cardíacos taquicárdicos, no galope, no tercer ruido, no soplos, pulsos llenos y llene capilar menor de dos segundos, frecuencia cardiaca: 120 latidos por minutos. Abdomen: suave, no distendido, no hepatoesplenomegalia, peristalsis presente. Sistema genitourinario: fimosis, dificultad al orinar, lesiones ampollares en pene y testículos incluyendo periné. Sistema nervioso central: Confusión. Nivel de conciencia por escala de coma de Glasgow 14/15. Tono, reflejos y fuerza normal. Pares craneales normales. No signos meníngeos. Durante los 10 primeros días presentó fiebre elevada que cedió al undécimo día.
Exámenes complementarios: Hemograma completo: Leucocitos: 2.4x 109/L.
Linfocitos: 31.6%. Monocitos: 18.4%. Neutrófilos: 50.0%.
eritrocitos: 4.90x109/L. Hb: 121 g/L. Plaquetas: 195x109/L.
Volumen corpuscular medio: 77.8 fL.
Hemocultivo: crecimiento de estafilococo aureus. VIH negativo.
El tratamiento incluyó: monitoreo de signos vitales permanente, hidratación parenteral y corrección de desequilibrio hidroelectrolítico, ropa de cama estéril y control de temperatura con calefacción local, cuidados de mucosa oral con solución salina estéril, cuidados de mucosa ocular con tetraciclina ungüento y oclusión, sonda nasogástrica para la alimentación, cura de lesiones en piel con sulfadiacina de plata y de las lesiones faciales y genitales con betadina, antiácidos orales, analgesia con petidina parenteral y paracetamol oral y antimicrobianos: los primeros siete días con ceftriaxone y oxacilina parenterales y siete días posteriores con eritromicina oral.
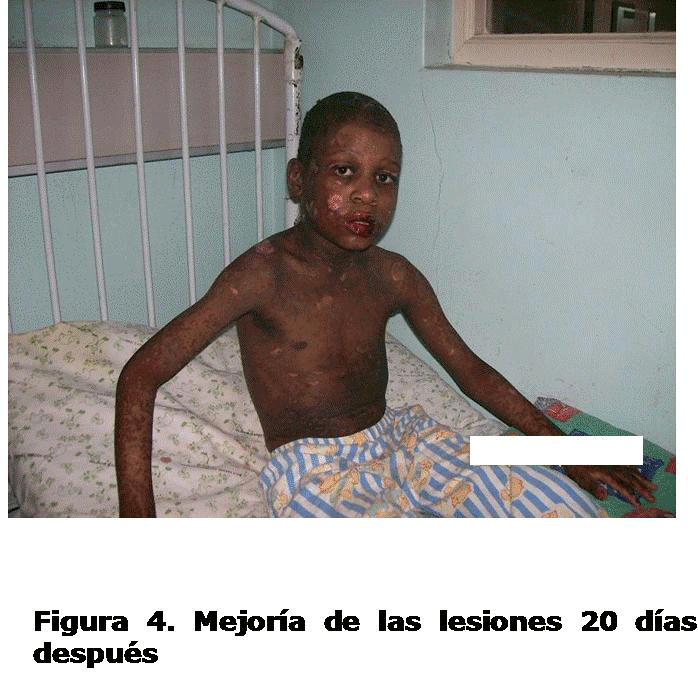
Con estas medidas se logró una evolución clínica favorable con mejoría de las lesiones en piel y mucosas, egresó el 3 de diciembre de 2007. (Figura 4)
DISCUSIÓN
Este caso coincide con la literatura revisada la cual plantea que generalmente aparece una conjuntivitis purulenta con uveítis y lesiones de piel que se rompen que dejan la piel desnuda, lo cual causa una importante pérdida de líquidos, anemia y un riesgo elevado de infección bacteriana sobreañadida y sepsis.1-3 Esta erupción puede estar precedida por una infección respiratoria inespecífica y puede ir acompañada de fiebre, decaimiento, escalofríos y malestar general.5, 9
El diagnóstico diferencial incluye el pénfigo ampollar, penfigoide, dermatosis lineal por IgA, erupción ampollar por drogas, síndrome de Reiter, síndrome de Kawasaki, síndrome de Behçet, vasculitis alérgicas y periarteritis nudosa.9-11
El tratamiento es sintomático y de sostén. Es muy importante el control oftalmológico pues es muy frecuente la pérdida de visión por úlceras corniales. Las lesiones orales pueden ser tratadas con lavados de la mucosa y glicerina. Las lesiones cutáneas pueden ser limpiadas con solución salina fisiológica. El uso de corticoesteroides es muy debatido, es el medicamento más utilizado en algunas unidades, pero otros autores plantean que incrementa la mortalidad por esta afección. Entre los tratamientos dirigidos a modificar la extensión del compromiso cutáneo el más controvertido es el uso de corticoesteroides sistémicos. Si bien se ha recomendado el uso de cursos cortos de estos fármacos, no se ha demostrado su eficacia en ensayos clínicos controlados. Algunos reportes de casos plantean el uso de plasmaféresis, ciclosporina, ciclofosfamida y anticuerpos monoclonales pero aún no se dispone de estudios concluyentes. Entre otros medidas se puede señalar correcto balance hidromineral, alimentación enteral, antimicrobianos sistémicos de amplio espectro, antitérmicos, correctas medidas de higiene y control ambiental.12-14
CONCLUSIONES
El síndrome de Steven Johnson es una enfermedad poco frecuente pero de una severa gravedad y su pronóstico depende de un diagnóstico precoz y un adecuado tratamiento.
REFERENCIAS BIBLIOGRÁFICAS
1.Gravante G, Delogu D, Marianetti M, Esposito G, Montone A. Toxic epidermal necrolysis and Steven Johnson syndrome: 11-years experience and outcome. Eur Rev Med Pharmacol Sci. 2007; 11(2):119-27.
2.Arroyo W, Luis E, Cheung C. Eritema multiforme secundario al uso de carbamazepina: Reporte de un caso. Rev med Costa Rica Centroam. 2009; 72(570):19-23.
3.Salami TA, Asalu AF, Samuel SO. Prevalence of cutaneous drug eruption in Nigerian with HIV/Aids. Niger Postgrade Med J. 2010; 17(2):160-3.
4.Mustafa N, Periyasamy P, Kamaruddin N. Steven Johnson syndrome in a patient with Cushing's disease. Med J Malaysia. 2009; 64(3):238-9.
5.Conforti R, Ferreira M, Abeldaño A. Síndrome de Stevens-Johnson: necrólisis epidérmica tóxica asociada a lamotrigina. Dermatol Argent. 2007; 13(3):190-4.
6.Pacheco L, Sánchez MA, Sánchez DL. Síndrome de Stevens-Johnson. Presentación de 1 caso. Rev Cubana Pediatr. 2001; 73(4):240-4.
7.Srinivasan R, Suchi ST. Cataract surgery in Steven Johnson syndrome. Indian J Ophthalmol. 2007; 55(6):483.
8.Gravante G, Delogu D, Marianetti M, Esposito G, Montone A. Toxic epidermal necrolysis and Steven-Johnson syndrome in oncologic patients. Eur Rev Med Pharmacol Sci. 2007; 11(4):269-74.
9.French LE, Trent JT, Kerdel FA. Use of intravenous immunoglobulin in toxic epidermal necrolysis and Stevens-Johnson syndrome: our current understanding. Int Immunopharmacol. 2006; 6(4):543-9.
10.Hernandez-Salazar A, Rosales PS, Rangel-Frausto S, Criollo E, Archer-Dubon C, Orozco-Topete R. Epidemiology of adverse cutaneous drug reactions. A prospective study in hospitalized patients. Arch Med Res. 2008; 37(7):899-902.
11.Sotozono C, Ang PKL, Koizumi N, Higashihara H, Ueta M, Inatomi T, et al. HLA class I and II gene polymorphisms in Stevens-Johnson syndrome with ocular complications in Japanese. Ophtalmology. 2007; 20(10):1-9.
12.Roujeau JC, Kelly JP, Naldi L, Rzany B, Stern RS, Anderson T, et al. Medication use and the risk of Stevens-Johnson syndrome or toxic epidermal necrolysis. N Engl J Med. 2005; 333(24):1600-7.
13.Teo L, Tay YK, Liu TT, Kwok C. Stevens-Johnson syndrome and toxic epidermal necrolysis: efficacy of intravenous immunoglobulin and a review of treatment options. Singapore Med J. 2009; 50(1):29-33.
14.Vasavada AR, Dholakia SA. Phacoemulsification in total white cataract with Stevens-Johnson syndrome. Indian J Ophthalmol. 2007; 55:146-8.
Recibido: 8 de diciembre de 2010
Aprobado: 4 de marzo de 2011
Dr. Ener de Jesús Fernández Brizuela. Email: fbener@finlay.cmw.sld.cu
