










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Archivo Médico de Camagüey
versión On-line ISSN 1025-0255
AMC vol.19 no.3 Camagüey mayo.-jun. 2015
CASOS CLÍNICOS
Síndrome de Stevens-Johnson: presentación de tres casos
Stevens-Johnson's syndrome: presentation of three cases
Dr. Jorge Luis Váquez Cedeño; Dr. Alejandro Vázquez Soto; Dr. Yosvanis Ortiz Jiménez; Dra. Mabel Brito Bartumeu; Dra. Laura Vázquez Brito
Hospital Universitario Héroes del Baire. Isla de la Juventud, Cuba.
RESUMEN
Fundamento: el Síndrome de Stevens-Johnson (SSJ) es una enfermedad grave, a menudo fatal, que ha sido considerada como un tipo de eritema multiforme, es causada generalmente por fármacos y de no ser diagnosticado y tratado de forma oportuna puede asociarse a secuelas importantes y la muerte.
Objetivo: describir y reseñar el tratamiento y evolución clínica del cuadro clínico de tres casos con diagnóstico de Síndrome de Stevens-Johnson, ingresados en Cuidados Intensivos.
Presentación de casos: se presentan tres enfermos, dos pertenecientes al sexo femenino y uno al masculino, con diagnóstico de SSJ asociados al empleo de fármacos. En los tres casos el tratamiento consistió en medidas de cuidados generales para la profilaxis y el tratamiento de complicaciones, esteroides sistémicos y además IgG intravenosa de producción nacional (intacglobin). Los tres pacientes evolucionaron de forma satisfactoria.
Resultados: se destaca la utilidad del uso de inmunoglobulina G intravenosa en el tratamiento de estos pacientes, así como su importancia en una unidad de cuidados para enfermos graves.
Conclusiones: en la casuística predominó el sexo femenino y el empleo precoz del Intacglobin (IgG IV) que contribuyó a una mejor evolución de los pacientes al detener la progresión de la enfermedad, evitar complicaciones y disminuir la estadía en las unidades de enfermos graves.
DeSC: SÍNDROME DE STEVENS-JOHNSON; INMUNOGLOBULINAS INTRAVENOSAS; INMUNOGLOBULINA G; ADULTO; INFORMES DE CASOS.
ABSTRACT
Background: Stevens-Johnson’s syndrome (SJS) is a serious disease, fatal most of the time, which has been considered as a type of erythema multiforme. It is generally caused by medicaments. If it is not diagnosed and treated at appropriate time it can be associated to considerable sequelae and death.
Objective: to describe the treatment and evolution of the clinical manifestations of three cases with the diagnosis of Stevens-Johnson’s syndrome admitted in the intensive care unit.
Cases presentation: the cases of two female patients and a male patient with the diagnosis of SJS, associated to the use of medicaments, are briefly presented. The treatment consisted of measures of general care for the prophylaxis and treatment of complications for the three cases. The patients were treated with intravenous IgG of national production (intacglobin) and systemic steroids. The three patients improved their condition satisfactorily.
Results: the use of intravenous immunoglobulin G in the treatment of these patients stands out; as well as the importance of the treatment of seriously ill patients in an intensive care unit.
Conclusions: female sex predominated in the casuistics. The early use of Intacglobin (IgG IV) contributed to a better improvement of the patient’s condition arresting the development of the disease, avoiding complications and decreasing the hospital stay of seriously ill patients.
DeSC: STEVENS-JOHNSON SYNDROME; IMMUNOGLOBULINS, INTRAVENOUS; IMMUNOGLOBULIN G; ADULT; CASE REPORTS.
INTRODUCCIÓN
El síndrome de Stevens-Johnson (SSJ) recibe su nombre por Albert Mason Stevens y Frank Chambliss Johnson, pediatras estadounidenses que en 1922 publicaron conjuntamente en el American Journal of Diseases, una descripción de dos pacientes con erupciones cutáneas generalizadas, fiebre continua, mucosa oral inflamada y conjuntivitis purulenta grave. 1-3
El síndrome de Stevens-Johnson se presenta con una incidencia anual de 1, 89 casos por millón de habitantes en los Estados Unidos, Alemania reporta una incidencia similar. 1 La condición es más común en adultos que en niños y las féminas se ven afectadas más a menudo que los hombres en una proporción de dos a uno (2:1). En la actualidad se define como la erosión severa de al menos dos superficies mucosas con necrosis extensa de los labios y boca y una conjuntivitis purulenta. El desprendimiento epidérmico puede ocurrir pero menos de un 10 % de la superficie corporal es afectada. Incluye también la aparición de fotofobia, deterioro visual y ceguera. 4
El SSJ, es una enfermedad grave aguda y a menudo fatal originada por una hipersensibilidad, que incluye como tejidos dianas la piel y las membranas mucosas y que en su forma más grave pone en peligro la vida del paciente. Puede ser inducida por muchos factores precipitantes, entre los cuales la toxicidad a fármacos es la más frecuente ejemplo de ellos son: sulfadiazina, sulfametoxazol-trimetoprim, penicilinas, sulfassalazina, ethambutol, isoniazida, quinolonas, cefalosporinas, tetraciclinas, antifungicos, fenobarbital, fenitoína, carbamazepina, valproato de sodio y antinflamatorios no esteroideos. Agentes infecciosos dentro de los que se encuentran bacterias como: C.Diphtheriae, brucela, microbacterias, tularemia, Streptococcus, Salmonella typhi, Mycoplasma; virus como: enteroviruses, adenoviruses, virus herpes simples, HIV, coxsackie, influenza, hepatitis, varicela y Epstein Bar. La literatura, además, describe casos por malaria y hongos. 1-4
La enfermedad puede tener un inicio repentino con manifestaciones inespecíficas y puede incluir síntomas tales como la fiebre, que es frecuente pudiendo llegar hasta 39 o 40 °C, cefalea, dolor de garganta y en la mucosa oral y malestar general, que en ocasiones se confunde con alguna otra enfermedad intercurrente. Es necesario destacar que estos síntomas suelen preceder a las manifestaciones cutáneas por unos días. Luego y de manera rápida los síntomas constitucionales se agravan, con taquicardia, postración y dolores articulares. La estomatitis es un síntoma característico con vesículas sobre los labios, lengua y mucosa bucal, se agrava con pseudomembranas, pérdida de sangre, salivación y ulceraciones que hacen difícil la alimentación o la ingestión de líquidos. 1-3, 5
Es frecuente que se desarrolle una conjuntivitis bilateral que puede ser catarral, purulenta e incluso pseudomembranosa pudiendo ocurrir úlceras en las córneas. Y pueden existir sinequia a nivel de los párpados. Puede además, aparecer frecuentemente epistaxis, rinitis, así como la formación de costras en los orificios nasales. La cara, manos y pies son también afectados e invadidos por una erupción hemorrágica, vesícula-ampollar o petequial, con inflamación de algunos o todos los orificios, boca, nariz, conjuntivas, uretra, vagina y ano. Se muestran lesiones diseminadas por el cuerpo y de existir vaginitis erosiva puede ser grave, lo que puede llegar a ocasionar sinequias con importantes secuelas. 6-8
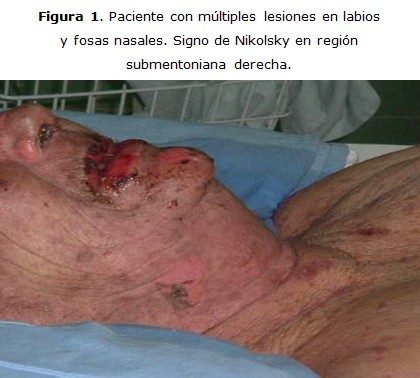
La lesión más característica en la piel es descrita como lesión en diana, puede estar presente el signo de Nikolsky, que consiste en un desprendimiento de la piel al pasar levemente el dedo. Aunque estas lesiones pueden aparecer en cualquier lugar, la cara, cuello y tórax son generalmente los más afectados. Las mucosas pueden presentar eritema, edema, ulceración y necrosis. 1, 6, 7, 9
Pueden existir trastornos gastrointestinales asociados. En las formas más graves de este proceso se pueden presentar además, artritis, convulsiones, coma, arritmias cardíacas, pericarditis; así como miositis, artritis, hepatopatías y sepsis generalizada. También se han descrito neumonía, deshidrataciones, alteraciones de los electrolitos y necrosis tubular aguda. La muerte sobreviene sin tratamiento en el 5 al 15% de los casos. El riesgo de recurrencia puede ser superior al 37 %.
En el diagnóstico diferencial se debe tener presente las siguientes enfermedades:
-. Pénfigo vulgar.
-. Pénfigo agudo febril.
-. Dermatitis herpetiforme de Duhring-Brock.
-. Epidermólisis bulosa hereditaria o pénfigo traumático.
-. Dermatitis exfoliativa de Ritter.
-. Necrolisis Tóxica epidérmica.
-. Acrodermatitis enteropática.
Los pilares en el tratamiento del paciente con SSJ son: 10-13
-. Medidas generales.
-. Cuidados especiales de la piel y mucosas.
-. Esteroides parenterales. Inmunoglobulina G intravenosa (intacglobin).
PRESENTACIÓN DE CASOS
Caso 1
Paciente J. I. S, masculino de 48 años de edad, mestizo, colaborador en la República Popular de Angola que ingresa en la Unidad de Cuidados Intensivos de la Clínica Meditex en Luanda con diagnóstico de Síndrome de Stevens-Johnson. Antecedentes de haber presentado un episodio de paludismo por lo que recibió tratamiento con fansidar, medicamento de la familia de las sulfas y 48 horas después aparecen lesiones eritematopapulosas a nivel de los labios, que se extiende con el paso de las horas al resto de la mucosa oral y a toda la piel afectando el tórax, abdomen, glúteos y extremidades. En cara, manos y pies las lesiones eran vesiculoampollar. No se presentaron lesiones en cuero cabelludo. Evolutivamente las lesiones confluyen formando grandes áreas de flictenas y costras. En el tratamiento se emplearon curas locales y esteroides (metilprednisolona en pulsos de 500 mg diarios) e inmunoglobulina G intravenosa (intacglobin) a razón de 400 mg/Kg/diario por cinco días. La evolución fue favorable con una estadía de una semana en Cuidados Intensivos, siendo egresado sin complicaciones.
Caso 2
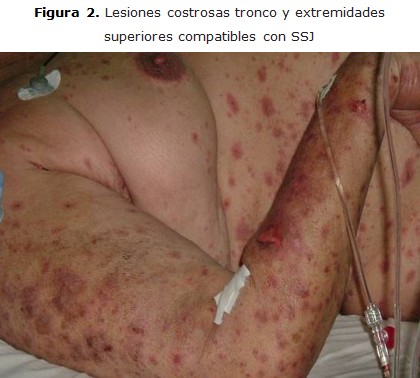
Paciente R.G.R, femenina de 83 años de edad y piel blanca, que ingresa en la sala de medicina del Hospital Héroes del Baire, en Nueva Gerona, con diagnóstico de dermatitis, dos días después de su ingreso se interconsulta con la guardia de Cuidados Intensivos, por la posibilidad de que la paciente presentara un Stevens-Johnson, lo que se confirma y es traslada a la unidad de Cuidados Intermedios. Se recoge en el interrogatorio que la enferma había presentado cinco días antes un cuadro de infección respiratoria, para lo cual se le había indicado cefalexina oral. Según refirió la paciente, ella no había ingerido ningún otro fármaco. Desarrolló lesiones eritematopapulosas en toda la piel, y presentó costras sangrantes a nivel de piel y mucosas, además desarrolló sinequia en los párpados, que obligó a realizar separación de los mismos casi diariamente para evitar su reproducción, no presentó ulceraciones corneales. En el tratamiento se utilizaron esteroides e intacglobin 400 mg/kg diario por cinco días, con lo cual se logró detener la progresión de las lesiones, lo que facilitó que la paciente evolucionara a la curación (figuras 1, 2).
Caso 3
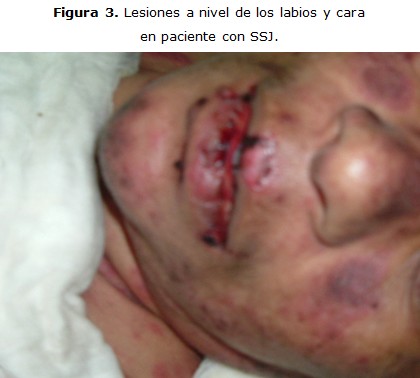
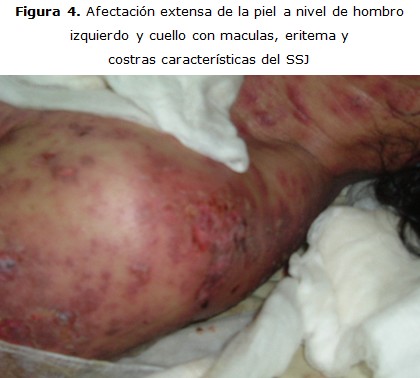
Paciente M. P. S. de 40 años, sexo femenino, raza mestiza, que ingresa en el Hospital Héroes del Baire, de Nueva Gerona con diagnóstico de Síndrome de Stevens-Johnson. Se trataba de una enferma con antecedentes de cuadro gripal, para lo cual se le indicó kogrip (paracetamol + dexclorferinamida). Tres días después comienzan a aparecer lesiones en la mucosa oral que se extienden a la piel en los días siguientes. Al momento de su ingreso ya presentaba lesiones eritematopapulosas diseminadas por toda la piel, observándose además lesiones costrosas sangrantes. El tratamiento incluyó además de los cuidados de la piel, esteroides e intacglobin. La evolución de la enferma fue positiva, al término de su curación fue egresada de la unidad de graves en siete días (figura 3, 4).
DISCUSIÓN
El SSJ es una enfermedad de presentación poco frecuente, secundaria a una reacción idiosincrática que en la mayoría de los casos se produce por medio de las drogas de uso frecuente como son: antibióticos, anticonvulsivantes y los antiinflamatorios no esteroideos, entre otros. Es precisamente el sustrato inmune lo que favorece que sea más común en el sexo femenino, como ocurrió en la presente investigación y que haya sido, además, señalado en varios trabajos revisados sobre el SSJ; otras causas incluyen infecciones bacterianas, virales u otras causas infecciosas. 3, 11, 14
En la anamnesis, con gran frecuencia, se relacionan como causa del SSJ, el uso de fármacos y es común que las primeras manifestaciones clínicas aparezcan entre cuatro a diez días luego del empleo de los mismos. Por otra parte, es usual que los síntomas y signos se confundan con manifestaciones clínicas de otras enfermedades, como ocurrió en uno de los pacientes de los casos expuestos, que fue ingresado con el diagnóstico de dermatitis. Los síntomas se iniciaron entre el segundo y quinto día de la exposición a fármacos con una media de 3, 3 días.
Dentro de los medicamentos que se documentaron en los tres casos se encontraron: antibióticos como el fansidar (que es una sulfa) y la cefalexina (cefalosporina de primera generación) y analgésicos como el paracetamol. Es de destacar que en dos pacientes se presentaron manifestaciones respiratorias, las que se interpretaron como infección del aparato respiratorio, pero en ninguno de los casos pudo ser aislado un germen responsable del cuadro respiratorio y su posible asociación al SSJ.
El SSJ puede manifestarse con un espectro de síntomas y signos muy variados, desde una enfermedad con pocas manifestaciones clínicas hasta un cuadro severo con peligro para la vida de los pacientes. Las lesiones a nivel de las mucosas se presentan en más del 90 % de los casos y son de gran valor en el diagnóstico, 15-17 como ocurrió en los tres casos presentados en este trabajo.
El tratamiento incluyó la retirada del fármaco que pudo haber desencadenado el SSJ, medidas generales y el empleo de IgG intravenosa (intacglobin) a dosis inmunosupresoras de 400 mg/kg/día por cinco días. En opinión de los autores, el uso del intacglobin fue decisivo en una menor estadía y en la evolución positiva de los casos, por lo que se recomienda su utilización como tratamiento de primera línea en los enfermos con SSJ, previa determinación de IgA, pues su déficit se asocia a anafilaxia en el tratamiento de esta enfermedad y de otras con sustrato inmunológico en las que se ha empleado la IgG intravenosa. En los casos en que no sea posible la determinación de los niveles de IgA, como sucedió en los tres casos presentados, se recomienda sea utilizado con precaución a un ritmo de administración lento y evaluar los riesgos y posibles beneficios.
En la investigación predominó el sexo femenino y se considera que el diagnóstico oportuno junto al empleo precoz del intacglobin (IgG IV) pudo haber contribuido en la evolución de los pacientes al detener la progresión de la enfermedad, evitar complicaciones y disminuir la estadía en las unidades de enfermos graves.
REFERENCIAS BIBLIOGRÁFICAS
1. Harr T, French LE. Toxic epidermal necrolysis and Stevens-Johnson Syndrome. Harr and French Orphanet Journal of Rare Diseases [Internet]. 2010 Dic [citado 24 Oct 2014];5(3):[aprox. 11 p.]. Disponible en: http://www.ojrd.com/content/5/1/39
2. David A, Wetter MD, Michael J, Camilleri MD. Clinical, Etiologic, and Histopathologic Features of Stevens-Johnson Syndrome During an 8-Year Period at Mayo Clinic. Mayo Clin Proc. 2010 Feb;85(2):131-8.
3. Pedigoni A, Domingues G, Penna H, Delascio R, Sandre L, Carlos A. Síndrome de Stevens-Johnson e Necrólise Epidérmica Tóxica em Medicina Intensiva. RBTI. 2006 Jul-Sep;18(3):292-7.
4. Ward KE, Archambault R, Mersfelder TL. Severe adverse skin reactions to nonsteroidal antiinflammatory drugs: A review of the literature. Am J Health Syst Pharm. 2010 Feb;67(3):206-13.
5. Gerull R, Nelle M, Schaible T. Toxic epidermal necrolysis and Stevens-Johnson syndrome: a review. Crit Care Med. 2011 Jun;39(6):1521-32.
6. Castana O, Rempelos G, Anagiotos G, Apostolopoulou C, Dimitrouli A, Alexakis D. Stevens-Johnson Syndrome: a case report. Anna Burns Fire Disast. 2009 Sep;22(3):147-51.
7. Quiñones J, Chávez JA, Bernárdez O. Síndrome de Stevens-Johnson: presentación de un caso. AMC. May-Jun 2011;15(3):576-84.
8. Chen J, Zeng Y, Xu H. High-dose intravenous immunoglobulins in the treatment of Stevens-Johnson syndrome and toxic epidermal necrolysis in Chinese patients: a retrospective study of 82 cases. Chinese. 2010 Nov-Dic;20(6):743-7.
9. Sawicki J, Ellis AK. Stevens-Johnson syndrome: A review of 14 adult cases with one fatal outcome. Anna Allerg Asth Immunol. 2013 Mar;110(3):207–209.e1.
10. Barron SJ, Del Vecchio MT, Aronoff SC. Intravenous immunoglobulin in the treatment of Stevens-Johnson syndrome and toxic epidermal necrolysis: a meta-analysis with meta-regression of observational studies. Int J Dermatol [Internet]. 2014 Abr [citado 24 Oct 2014];[aprox. 1 p.]. Disponible en: http://onlinelibrary.wiley.com/doi/10.1111/ijd.12423/full
11. Pozzo-Magana BR, Lazo-Langner A, Carleton B, Castro-Pastrana L, Rieder M. A systematic review of treatment of drug-induced stevensjohnson Syndrome and toxic epidermal necrolysis in children. J Popul Ther Clin Pharmacol. 2011 Mar;18(1):e121-e133.
12. Sawicki J, Ellis AK. Stevens-Johnson syndrome: A review of 14 adult cases with one fatal outcome. Anna Allerg Asth Immunol. 2013 Mar;110(3):207–209.e1.
13. Campagna C, Tassinari D, Neri I, Bernardi F. Mycoplasma pneumoniae-induced recurrent Stevens-Johnson syndrome in children: a case report. Pediatr Dermatol. 2013 Sep-Oct;30(5):624-6.
14. Wetter D, Camilleri M. Clinical, Etiologic, and Histopathologic Features of Stevens-Johnson Syndrome During an 8-Year Period at Mayo. Clin Proc. 2010 Feb;85(2):131-8.
15. Sawicki J, Ellis AK. Stevens-Johnson syndrome: A review of 14 adult cases with one fatal outcome. Annals of Allergy, Asthma & Immunology. 2013 marzo; 110(3):207–209.e1.
16. Campagna C, Tassinari D, Neri I, Bernardi F. Mycoplasma pneumoniae-induced recurrent Stevens-Johnson syndrome in children: a case report. Pediatr Dermatol. 2013 Sep-Oct;30(5):624-6.
17. Wetter D, Camilleri M. Clinical, Etiologic, and Histopathologic Features of Stevens-Johnson Syndrome During an 8-Year Period at Mayo Clinic. Mayo Clin Proc. 2010 Feb;85(2):131-138.
Recibido: 5 de diciembre de 2014
Aprobado: 26 de marzo de 2015
Dr. Jorge Luis Váquez Cedeño. Especialista de II Grado en Medicina Interna y II Grado Medicina Intensiva y Emergencias. Máster en Urgencias Médicas. Profesor Auxiliar. Hospital Universitario Héroes del Baire. Isla de la Juventud, Cuba. Email: jlvazquez@infomed.sld.cu

