










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Archivo Médico de Camagüey
versión On-line ISSN 1025-0255
AMC vol.19 no.3 Camagüey mayo.-jun. 2015
CASOS CLÍNICOS
Síndrome del QT largo y muerte súbita cardiovascular
Long QT syndrome and sudden cardiovascular death
Dr. Fernando Carreras Calvo I; Dr. Rolando Castellanos Rojas II; Dra. Rut Perozo Panicello III; Dr. Lázaro Ramírez Lana II
I Hospital Universitario Martín CHang Puga. Camagüey, Cuba.
II Hospital Universitario Manuel Ascunce Domenech. Camagüey, Cuba.
III Policlínica Universitario Francisco Peña Peña. Camagüey, Cuba.
RESUMEN
Fundamento: el síndrome de QT largo es una canalopatía arritmogénica, caracterizada por una grave alteración en la repolarización ventricular, traducida electrocardiográficamente por una prolongación del intervalo QT, que predispone a la muerte súbita por arritmias ventriculares malignas, del tipo torsada de punta.
Objetivo: presentar un caso de síndrome QT poco frecuente en nuestro medio.
Caso Clínico: paciente de 31 años de edad con antecedente de salud, que ingresó en dos ocasiones durante su embarazo por cifras elevadas de tensión arterial en el año 2013. Presentó varios cuadros de sincope del cual se recuperaba espontáneamente. Ingresó en enero de 2014 por trastornos dispépticos y epigastralgia, al estar hospitalizada hizo un cuadro de sincope y taquicardia ventricular documentadas por electrocardiograma, donde llegó hacer torsada de punta y parada cardiorrespiratoria, por lo que fue necesario la reanimación cardiorrespiratoria y entubación endotraqueal, así se mantuvo 48 horas y salió de este cuadro. Se trasladó al instituto de cardiología en ciudad de la habana, donde fue estudiada por el equipo de arritmias y se decidió la implantación de un dispositivo de desfibrilación automático implantable.
DeCS: SÍNDROME DE QT PROLONGADO; CANALOPATÍAS; MUERTE SÚBITA CARDÍACA; ADULTO; INFORMES DE CASOS.
ABSTRACT
Background: long QT syndrome is an arrhythmogenic canalopathy characterized by a serious alteration in the ventricular repolarization, translated electrocardiographically by a prolongation of the QT interval that predispose to sudden death caused by malignant ventricular arrhythmias tracing in torsades de pointes.
Objective: to present the clinical case of a patient with this syndrome that is infrequent in our environment.
Clinical case: a thirty-one-year-old patient with antecedents of being a healthy person, who was admitted in the hospital twice during her pregnancy because of high arterial pressure in 2013. The patient presented syncope manifestations from which she recovered spontaneously. In January 2014, the patient is admitted in the hospital with dyspeptic disorders and epigastralgia. After being admitted, the patient presented syncope manifestations and ventricular tachycardia exhibited on the electrocardiogram tracing in torsades de pointes and cardiac arrest. It was necessary to perform a cardiopulmonary resuscitation and endotracheal intubation. The patient presented this combination of manifestations for 48 hours. She is sent to the Cardiology Institute of Havana where she is studied by the arrhythmia medical team and the implantation of an implantable cardioverter-defibrillator is decided.
DeCS: LONG QT SYNDROME; CHANNELOPATHIES; DEATH, SUDDEN, CARDIAC; ADULT; CASE REPORTS.
INTRODUCCIÓN
Las enfermedades cardiovasculares genéticas sin cardiopatía estructural, son enfermedades de baja prevalencia, asociadas a mayor riesgo de arritmias ventriculares graves, sincope y muerte súbita. 1
El síndrome de QT largo (SQTL) es una enfermedad que se caracteriza por prolongación del tiempo de repolarización ventricular y predisposición a arritmias ventriculares potencialmente letales. La causa del SQTL puede ser congénita o adquirida. La forma congénita de este síndrome suele ser de origen genético, el adquirido es generado principalmente por la acción de fármacos que prolongan la duración del potencial de acción a expensas de la inhibición de los canales del sodio o del potasio y por trastornos hidroelectrolíticos, como hipopotasemia, hipomagnesemia e hipocalcemia, los cuales generan una alta predisposición a presentar arritmias ventriculares malignas como la torsada de punta. 2
Aproximadamente un 10-20 % de las muertes súbitas cardíacas, son producidas por enfermedades sin cardiopatía estructural, conocidas como canalopatías o enfermedades eléctricas primarias, dentro de las cuales las más representativas son: síndrome de QT largo congénito o adquirido, síndrome de Brugada, síndrome de QT corto congénito y la taquicardia ventricular polimorfa catecolaminérgica. Estas canalopatías pueden presentar superposición de los fenotipos clínicos y electrocardiográficos.
El electrocardiograma (ECG) es de suma importancia para el diagnóstico, dado que estas mutaciones genéticas solo se encuentran en un porcentaje reducido de casos. La prevalencia de estas enfermaddes es muy baja y por consiguiente, no existen resultados concluyentes basados en parámetros clínicos o genéticos, provenientes de grandes estudios prospectivos y randomizados. Por lo tanto, la estratificación del riesgo está basada en datos retrospectivos de registros multicéntricos. 3
En la actualidad se han descrito hasta 10 tipos del síndrome, si bien los tipos uno, dos y tres, engloban la gran mayoría de los casos, siendo el tipo uno el más prevalente. La administración de β-bloqueantes se considera el tratamiento inicial de los pacientes con diagnóstico confirmado, y la reducción de eventos es especialmente pronunciada en los tipos uno y dos, en los que debido al defecto genético causal (mutaciones en la subunidad α del canal de potasio) los eventos se relacionan con las situaciones de estrés o actividad física. 4
El objetivo de este trabajo es presentar un caso clínico de este síndrome, poco frecuente.
CASO CLÍNICO
Paciente de 31 años de edad con antecedente de salud, que ingresó en dos ocasiones durante su embarazo, por cifras elevadas de tensión arterial en el año 2013. Presentó varios cuadros de sincope, del cual se recuperaba espontáneamente. Ingresó en enero de 2014 por trastornos dispépticos y epigastralgia, y al estar hospitalizada hizo un cuadro de sincope y taquicardia ventricular documentadas por electrocardiograma, donde llegó hacer fibrilación ventricular, torsada de punta y parada cardiorrespiratoria, por lo que es necesario la reanimación cardiorrespiratoria y entubación endotraqueal, así se mantuvó 48 horas y salió de este cuadro.
Después de ser valorada por los intensivistas del hospital deciden administrar amiodarona, al inicio, por vía endovenosa y luego se pasa a la vía oral y se solicita valoración por cardiología; 24 horas después el cardiólogo determina que la paciente presenta un síndrome de QT largo, se retira el uso de la amiodarona y comienza tratamiento con propanolol de 10mg cada 8 horas; 48 horas más tarde, la paciente es trasladada al instituto de cardiología en ciudad de la habana, es estudiada por el equipo de arritmias, confirmándose el diagnóstico y se decide la implantación de un dispositivo de desfibrilación automático implantable.
Electrocardiograma: Figura 1, 2, 3 y 4.
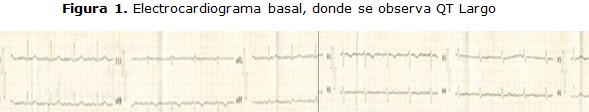
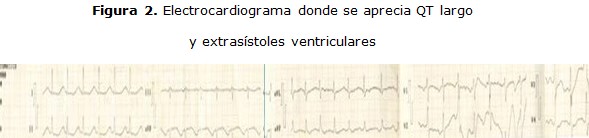


RX Tórax. Normal
Ecocardiograma. Normal
DISCUSIÓN
El SQTL es una alteración cardíaca causada por un alargamiento de la fase derepolarización del potencial de acción ventricular. Está causado por una disfunción de la actividad de los ionóforos de potasio (K), durante esta fase. La despolarización normal se produce por una entrada rápida de cargas positivas, en concreto iones sodio (Na) y Calcio (Ca), al interior de la célula miocárdica. 5 La reducción de cargas positivas que inicia la repolarización, se produce cuando la salida de iones K supera la entrada de Na y Ca mencionada. Cuando esta salida de K se produce de forma deficiente o lenta, se alarga la repolarización lo cual se constata en el ECG con el alargamiento del QT.
El SQTL implica un aumento del riesgo de arritmia ventricular grave. Puede ser congénito por una canalopatía o adquirido, generalmente producido por un fármaco, y en este caso se denomina únicamente QT largo (QTL). 5, 6 Los antidepresivos juegan un papel importante provocando alargamiento del QT y arritmias letales. 7-10 Al igual que la cardiopatía isquémica. 11
La clasificación utilizada en el pasado se basa en la presentación homocigota o heterocigota de la enfermedad, que dan lugar a los síndromes de Jervell-Lange-Nielsen (con sordera) y Romano Ward (sin sordera), respectivamente. 11 La clasificación actual enfatiza los hallazgos genéticos. En 1995-1996 se describieron los 3 principales genes asociados con la enfermedad que codifican para unidades formadoras del poro de los canales de potasio (IKs e IKr) y de sodio (Na v 1, 5); explican cerca del 65 % de los casos. Si bien en los años subsecuentes se han añadido siete genes más, a la lista, éstos explican tan sólo cerca del 5 % de los casos. 12
Los canales iónicos son proteínas transmembranales encargadas de transportar iones a través de la membrana celular; los canales implicados en el SQTL son selectivos o especializados en el transporte de un solo ion y dependientes de voltaje, es decir, su activación ocurre a determinado voltaje intracelular (varía según el subtipo de canal). Los fenómenos eléctricos y contráctiles que suceden en el cardiomiocito son controlados por estas estructuras.
Los canales iónicos forman complejos macromoleculares; hay una unidad principal formadora del poro del canal y proteínas auxiliares que lo regulan. La alteración en la unidad formadora del poro, conocida como alfa, genera los 3 subtipos más comunes de SQTL: SQTL1 (afección en el canal de potasio (IKs), SQTL2 (afección en el canal de potasio (IKr) y SQTL3 (afección en el canal de sodio). Al ser los más frecuentes, han sido mejor caracterizados clínica y genéticamente. 13
El síndrome llamado Jervell-Lange-Nielsen corresponde en la actualidad a las variedades de SQTL 1 y 5. De manera característica, los pacientes cursan con sordera congénita y tienen mutaciones homocigotas o heterocigotas compuestas, que afectan a la corriente IKs. El síndrome de Romano Ward abarca desde la variedad SQTL 1 hasta la 10 y no cursa con sordera. 14
El síndrome de QT largo tipo 1 (SQTL1) los pacientes suelen presentar episodios de arritmia ventricular al realizar ejercicio o al estimular el simpático (68 %); la natación se ha descrito como un deporte disparador de arritmias en el SQTL1. 8 Es el subtipo más frecuente y explica 30-35 % de los casos. El gen afectado es el KvLQT1 (o KCNQ1), localizado en el cromosoma 11 (11p15.5), codifica la subunidad α, del canal de potasio IKs. El potencial de acción se prolonga por una disminución de la corriente saliente de potasio (K+) durante fase 3 del potencial de acción.
En el síndrome de QT largo tipo 2 (SQTL2), suelen presentan arritmias ventriculares en respuesta al estrés emocional (49 %) o estímulos auditivos súbitos por ejemplo, al reloj despertador y con menos frecuencia durante el sueño o el ejercicio. Este subtipo es particularmente susceptible a presentar arritmias en el período posparto. La penetrancia estimada es del 79 % y significa que hasta un 20 % de los casos pueden tener un ECG no diagnóstico. En el SQTL2, la onda T suele ser de baja amplitud, bífida, con muescas. El gen afectado es el KCNH2 o HERG, localizado en el cromosoma 7, el cual codifica la subunidad α del canal de potasio (IKr); explica 25-30 % de los casos. 9 La disfunción de este canal disminuye la corriente saliente de K+ durante la fase 3 del potencial de acción, prolongando su duración.
Síndrome de QT largo tipo 3 (SQTL3) tienen un riesgo mayor de presentar arritmias malignas durante el reposo (sueño) o bradicardia. 11 La penetrancia de las mutaciones en el gen SCN5A es cercana al 90 %. El ECG en el SQTL3 suele mostrar onda T acuminada, de aparición tardía, que deja observar con claridad el alargamiento del segmento ST. Estos pacientes suelen ser menos sintomáticos que los casos con SQTL1 o SQTL2, pero los eventos son característicamente más letales. El gen afectado en el SQTL3 es el SCN5A, que codifica para la subunidad α del canal de sodio localizado en el cromosoma 3 (3p21-24); es causante de la enfermedad en el 5-10 % de los casos. La inactivación defectuosa del canal permite la entrada sostenida de sodio (Na+) durante la fase 2 del potencial de acción y prolonga su duración.
Síndrome de QT largo tipo 4 es una variedad rara y explica cerca del 1 % de los casos. Condiciona un SQTL muy atípico con un gran espectro de arritmias que incluyen taquicardia ventricular polimórfica catecolaminérgica, fibrilación auricular, trastornos en la conducción intraventricular, disfunción del nódulo sinusal y bradicardia; con frecuencia, los casos tienen incluso el QT corregido (QTc) en los límites normales. El gen afectado es el ANKB, localizado en el cromosoma 4 (4q25-27), el cual codifica la síntesis de anquirina-β, una proteína estructural que vincula proteínas de la membrana del cardiomiocito con proteínas del citoesqueleto. Estas proteínas son: la bomba de sodio y potasio, el intercambiador sodio – calcio (Na/Ca) y el receptor a inositol trifosfato (InsP3R). Las mutaciones que causan pérdida de la función de anquirina-β, resultan en un incremento de la concentración de calcio intracelular, así como en una alteración en la expresión de N/K ATP-asa y en el intercambiador Na/Ca. La elevación de las concentraciones de calcio da lugar a post-despolarizaciones tempranas y tardías. De esta manera, las arritmias ventriculares observadas en las mutaciones del gen de anquirina-β se deben a despolarizaciones espontáneas generalmente en respuesta a la estimulación catecolaminérgica. 12
Síndrome de QT largo tipo 5 (SQTL5) está condicionado por cambios de secuencia del gen KCNE1 localizado en el cromosoma 21 (21q22.1-p22). Codifica la síntesis de la subunidad β del canal IKs, conocida también como subunidad minK que regula al canal IKs. Explica menos del 1% de los casos. 11
Síndrome de QT largo tipo 6 (SQTL6) el gen afectado es el KCNE2 localizado en el cromosoma 21 (21q22.1). Codifica la subunidad β del canal de potasio, conocida también como subunidad MiRP1. 13
Síndrome de QT largo tipo 7 o Andersen-Tawil (SQTL7) los hallazgos dismórficos y las alteraciones electrocardiográficas de este síndrome fueron descritos por primera vez en 1971 por el Dr. Andersen y recapitulados en 1994 por el Dr. Tawil, citado por Van den Branden B, et al, 1 pero la descripción genético-molecular se publicó apenas en el año 2001.
Síndrome de QT largo tipo 8 (SQTL8) resulta de mutaciones en el gen CACNA1que codifica el canal de calcio tipo L Cav 1.2, localizado en el cromosoma 12 (12p13.3). Ocasiona el síndrome de Timothy, caracterizado por malformaciones cardiacas, deficiencia inmunológica, hipoglucemia intermitente, trastornos cognitivos incluido el autismo, fusiones interdigitales y QT largo que predispone a arritmias cardiacas y muerte súbita. 7Explica menos del 0,5 % de los casos.
Síndrome de QT largo tipo 9 (SQTL9) esta variedad resulta de mutaciones en el gen CAV3, localizado en el cromosoma 3 (3p25), que codifica la síntesis de caveolina. 3 La caveola es una invaginación de la membrana plasmática implicada en la endocitosis, la homeostasis de lípidos y la transducción de señales. Un importante componente de esta estructura es la caveolina, de la cual se conocen 3 subtipos; el subtipo 3 es específico de músculo esquelético y cardiaco. Algunos canales iónicos se colocalizan en la caveola, incluida la isoforma cardiaca de canal de sodio Nav1.5; recientemente se describieron diversas mutaciones en esta proteína que alteran las propiedades biofísicas del canal de sodio Nav1, 5 in vitro, generando un fenotipo similar al observado en el SQTL3. Se estima que explica < 1 % de los casos. 4
Síndrome de QT largo tipo 10 (SQTL10) esta variedad fue notificada en un caso muy grave con un QTc mayor de 600 ms, bradicardia fetal y bloqueo auriculoventricular (AV) 2 ƒ 1. Resulta de mutaciones en el gen SCN4B, localizado en el cromosoma 11 (11q23) que codifica para la subunidad β 4 de canal de sodio. Se han descrito 4 distintos subtipos de subunidades β que interaccionan y regulan las diversas isoformas de canal de sodio, pero sólo el subtipo 4 se ha asociado hasta ahora con arritmogénesis. 12 La incidencia de mutaciones en este gen no ha sido explorada, pero se estima menos de 1 %. 15-18
El SQTL se diagnostica por las manifestaciones clínicas como mareos, síncopes recurrentes, paro cardíaco y muerte súbita (MS) y la herramienta clave es el ECG con la medición del QT, el QT corregido (QTc) con la frecuencia cardíaca (FC) y los cambios morfológicos del segmento ST y la onda T. Los antecedentes familiares de paro cardíaco o MS, pueden contribuir a orientar el diagnóstico. El test genético, imposible de realizar hasta el momento en el nuestro país, permite confirmar el diagnóstico sobre todo en el QTL sin QTL, identificar cada subtipo y estratificar el riesgo. 19-21
La medición del QT debe realizarse con el registro a 25 mm/s y amplitud de 10 mm/mv, en las derivaciones II, V5 o V6, desde el inicio del QRS hasta el final de la onda T, en una media de 3 a 5 latidos. 22 El final de la T se define en el lugar donde su rama descendente cruza la línea isoeléctrica; su sitio de terminación puede ser difícil de determinar cuando existen T bifásicas, aplanadas o se asocian ondas U; estas últimas no deben ser incluidas en la medición, a menos que se encuentran fusionadas a la T o forman parte de su porción terminal. 23
Las variaciones en la anchura del QRS, como sucede con los bloqueos de rama y uso fármacos antiarrítmicos, modifican la despolarización y pueden también modificar la repolarización, lo que hace inexacta la medición del QT. El cálculo del punto J y T (JT) desde la S al final de la T pudiera ser la solución, pero estos valores no están bien establecidos. 24 La variabilidad del R-R y arritmias como la fibrilación auricular también pueden entorpecer el diagnóstico, por lo que debe realizarse la medición como mínimo en 10 complejos.
Los valores normales del QTc oscilan entre 350 y 450 milisegundos (ms) en los hombres y entre 360 y 460 ms en las mujeres. Las cifras superiores a 460 ms en los niños y jóvenes de 1 a 15 años, a 450 ms en los hombres y a 470 ms en las mujeres se consideran prolongadas, 25-27pero valores intermedios entre 430 ms y 470 ms pueden encontrarse tanto en sujetos sanos como en pacientes con SQTL, resultado de penetración variable, posibilidades de solapamiento, superposición, variabilidad de la duración del QT y dificultades en las mediciones. El QTc 470 ms en los hombres y a 480 ms en las mujeres, debe considerarse como prolongado, aún en pacientes asintomáticos sin historia familiar. No es posible determinar el valor exacto de QTc que desencadenará una torsada de punta (TDP), pero el riesgo aumenta en la medida que se prolonga este intervalo. Para definir el tratamiento debe descartarse el uso de fármacos, trastornos electrolíticos (hipopotasemia, hipomagnesemia), bradicardias, pausas, disfunción hepática o renal u otros factores que pudieran justificar la prolongación adquirida del QTc, en asociación o no con el SQTL congénito. 28
Los bloqueadores beta constituyen la primera línea de tratamiento, con una respuesta favorable en el SQTL1 y SQTL2; en el LQT3 pueden ser inefectivos, incluso tener un efecto perjudicial. 29
Los suplementos de potasio y la espironolactona, también pueden ser útiles sobre todo en el SQTL2. 30
La denervación simpática izquierda ha demostrado resultados favorables en el SQTL1 refractario a los bloqueadores beta. 31
El uso de bloqueadores de los canales de Na como la mexiletina, flecainide y ranolazina pueden tener un efecto beneficioso en el SQTL3, pero su uso sin la orientación genética pudiera ser dañino, en caso de asociación con otras entidades como el síndrome de Brugada. 32
El verapamilo, que es también un inhibidor de las corrientes tardías de Na+, pudiera reducir el QTL. 33
Los marcapasos cardíacos son efectivos en las TV pausa-dependientes (intervalos corto-largo-corto), bradicardias o bloqueos aurículo ventriculares con o sin prolongación del intervalo QTc. Tienen mayor utilidad en el SQTL3. Su asociación con bloqueadores beta puede ser útil en los pacientes con alto riesgo. 34
REFERENCIAS BIBLIOGRÁFICAS
1. Van den Branden B, Wever E, Boersma L. Torsade de pointes with short coupling interval. Acta Cardiol. 2010;65(3):345-6.
2. Smith DS. Is long QT síndrome a disease of abnormal mechanical contraction. Circ Arrhythm Electrophysiol. 2010;122(8):1353-54.
3. De Jesús Pérez VA, Haddad F, Vagelos RH, Fearon W, Feinstein J, Zamanian RT. Angina associated with left main coronary artery compression in pulmonary hypertension. J Heart Lung Transplant. 2009;28(5):527-30.
4. Tseng PT, Lee Y, Lin YE, Lin PY. Low-dose escitalopram for 2 days associated with corrected QT interval prolongation in a middle-aged woman: A case report and literature review. Gen Hosp Psychiatry. 2012;34(210):13-15.
5. Deshmukh A, Ulveling K, Alla V, Abuissa H, Airey K. Prolonged QTc interval and torsades de pointes induced by citalopram. Tex Heart Inst J. 2012;39(1):68-70.
6. FDA Drug Safety Comunication [Internet]. 2012 Mar 28 [citado 23 Abr 2014]. Disponible en: http://www.fda.gov/Drugs/DrugsSafety/ucm297391.htm
7. Howland RH. A critical evaluation of the cardiac toxicity of citalopram: Part1. J Psychosoc Nurs Ment Health Serv. 2011;49(2):13-6.
8. Van Gorp F, Whyte IM, Isbister GK. Clinical and ECG effects of escitalopram overdose. Ann Emerg Med. 2009;54(8):404-8.
9. Castro Ferman VM, Clements CC, Murphy SN, Gainer VS, Fava M, Weilburg JB, et al. QT interval and antidepressant use: A cross sectional study of electronichealth records. BMJ. 2013;29(1):346.
10. Zivin K, Pfeiffer PN, Bohnert AS, Ganoczy D, Blow FC, Nallamothu BK, et al. Evaluation of the FDA warning against prescribing citalopram at doses exceeding 40 mg. Am J Psychiatry. 2013;170(6):642-50.
11. Jiménez Candil J, González Matas JM, Cruz González I, Hernández Hernández J, Martín Diaz A, Pabón Andres P, et al. Pronóstico hospitalario del síndrome Coronario agudo sin elevación Del segmento ST determinado por una nueva escala de riesgo integrada por variables electrocardiográficas obtenidas al ingreso. Rev Esp Cardiol. 2010;63(7):851-5.
12. Yilmaz MB, Yontar C, Erdem A, Karadas F, Yalta K, Turgut OO, et al. Comparative effects of levosimendan and dobutamine on right ventricular function in patients with biventricular heart failure. Heart Vessels. 2009;24(5):16-21.
13. Wong JA, Gula LJ, Klein GJ, Yee R, Skanes AC, Krahn AD. Utility of tradmilltesting in identification and genotype prediction in Long-QT Syndrome. Circ Arrhythm Electrophydiol. 2010;3(3):120-25.
14. Drezner JA, Ackerman MJ, Cannon BC, Corrado D, Heidbuchel H, Prutkin JM, et al. Abnormal electrocardiographic findings in athletes: recognising changes suggestive of primary electrical disease. Br J Sports Med. 2013;47(3):153-67.
15. Chattha IS, Sy RW, Yee R, Gula LJ, Snakes AC, Klein GJ, et al. Utility of the 16-Shimizu W. Diagnosis Evaluation of Long QT Syndrome. Card Electroph Clin. 2012;4(8):29-37.
16. Viskin S, Belhassen B, Wilde AA. Irreplaceable antiarrhythmic medications are disappearing: the case of Quinidine. Heart Rhythm. 2010;7(1):863.
17. Haissaguerre M, Sacher F, Nogami A, Komiya N, Bernard A, Probst V, et al. Characteristics of recurrent ventricular fibrillation associated with inferolateral early repolarization. J Am Coll Cardiol. 2009;53(3):612-9.
18. Antzelevitch C, Yan GX. J wave syndromes. Heart Rhythm. 2010;7:549-58.
19. Viskin S. Idiopathic ventricular fibrillation. Le síndrome d Haissaguerre and the fear of J waves. J Am Coll Cardiol. 2009;53(4):620-2.
20. Ghosh S, Cooper DH, Vijayakumar R, Zhang J, Pollak S, Haissaguerre M, et al. Early repolarization associated with sudden death: insights from noninvasive electrocardiographic imaging. Heart Rhythm. 2010;7(5):534-7.
21. Márquez Diaz MF. El síndrome de QT largo: una breve revisión del diagnóstico electrocardiográfco incluyendo la prueba de Viskin. Arch Cardiol Mex. 2012;82(3):243-7.
22. Jiang W, Velazquez Guedez EJ, Kuchibhatla M, Samad Z, Boyle SH, Kuhn C, et al. Effect of escitalopram on mental stressinduced myocardial ischemia:Results of the REMIT trial. JAMA. 2013;309(1):2139-49.
23. Venetucci L, Denegri M, Napolitano C, Priori SG. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat Rev Cardiol. 2012;9(2):561–75.
24. Van der Werf C, Nederend I, Hofman N. Familial evaluation in catecholaminergic polymorphic ventricular tachycardia: disease penetrance and expression in cardiac ryanodine receptor mutation-carrying relatives. Circ Arrhythm Electrophysiol. 2012;5(6):748–56.
25. Watanabe H, Chopra N, Laver D. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009;15:380–3.
26. Zayas molina R. Actualización sobre el síndrome de QT largo congénito. Rev Cubana Invest Bioméd. 2012;31(2):34-56.
27. Chávez González E. El Intervalo QT, su origen e importancia del conocimiento de fórmulas para su medición en diferentes circunstancias clínicas. Cor Salud. 2014;6(1):34-40.
28. Hong RA, Jittirat A, Choi JJ. Flecainide suppresses de?brillator-induced storming in catecholaminergic polymorphic ventricular tachycardia. Pacing Clin Electrophysiol. 2012;359(3):794–7.
29. Kimura H, Zhou J, Kawamura M. Phenotype variability in patients carrying KCNJ2 mutations. Circ Cardiovasc Genet. 2012;5:344–53.
30. Faggioni M, Knollmann BC. Calsequestrin 2 and arrhythmias. Am J Physiol Heart Circ Physiol. 2012;302(3):H1250–H1256.
31. Priori SG, Chen SR. Inherited dysfunction of sarcoplasmic reticulum Ca2 þ handling and arrhythmogenesis. Circ Res. 2011;108(1):871–83.
32. Ruesta Casote V. Una forma atípica de síndrome de QT largo: Heraldos de muerte súbita cardíaca. Gac Méd Caracas [Internet]. Mar 2008 [citado 12 Jun 2014];116(1):[aprox. 6 p.]. Disponible en: http://www.scielo.org.ve/scielo.php?script=sci_arttext 47622008000100007
33. Características clínicas, pronóstico vital y funcional de los pacientes supervivientes a una muerte súbita extrahospitalaria ingresados en cinco unidades de cuidados intensivos cardiológicos. Rev Esp Cardiol. 2013;66(2):623-8.
34. Ochoa Montes LA, González Lugo M. Epidemiología de la muerte súbita cardíaca. Rev Cubana Hig Epidemiol. 2012;50(1):23-34.
Recibido: 29 de septiembre de 2014
Aprobado: 7 de abril de 2015
Dr. Fernando Carreras Calvo. Especialista de I Grado en Cardiología. Hospital Universitario Martín CHang Puga. Nuevitas, Camagüey, Cuba.

