










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Archivo Médico de Camagüey
versión On-line ISSN 1025-0255
AMC vol.19 no.5 Camagüey sep.-oct. 2015
CASOS CLÍNICOS
Osteopetrosis marmórea: reporte de un caso con esta rara enfermedad
Marble osteopetrosis: report of a case with this rare disease
Dra. Tania Cristobo Bravo; Dr. Sergio Rodríguez Télles; Dr. Gaetano Di Vasto Cuellar; Lic. Niurka González Cuesta; Dra. Idalia Morell Amarales
Hospital Pediátrico Universitario Dr. Eduardo Agramonte Piña. Camagüey, Cuba.
RESUMEN
Fundamento: la Osteopetrosis agrupa un conjunto de enfermedades óseas caracterizadas por aumento de la densidad ósea, debido a una disfunción osteoclástica. Se la conoce también como enfermedad marmórea o de Albers-Schönberg.
Objetivo: realizar la presentación de un caso de osteopetrosis marmórea y la revisión del tema.
Caso clínico: paciente de sexo masculino, de 14 años de edad. Sin antecedentes de consanguinidad de los padres, que acude por dolor óseo generalizado, que se irradiaba a lo largo de las extremidades, además de cefalea intensa que motivó estudio por neurología. En los estudios neurológicos resaltó la región mastoidea izquierda, por lo que se niega inflamaciones articulares, fiebre, erupción, nódulos, lesiones de piel. No existen elementos de hipoplasia medular asociada a la enfermedad en este paciente. En el examen físico actual no se evidencia hallazgo alguno de enfermedad inflamatoria crónica. Radiografías del paciente revelan un aumento generalizado de la densidad ósea, deformidades por desalineación, esclerosis ósea difusa, esto se traduce en huesos densos, anchos, sin canal medular, lo que representa cartílago hipermineralizado.
Conclusiones: los estudios radiológicos fueron positivos y confirmaron el diagnóstico de la Osteopetrosis marmórea en la forma benigna; no es necesario el uso de otros estudios de imágenes para confirmarlo.
DeCS: OSTEOPETROSIS/radiografía; ESCLEROSIS; RESORCIÓN ÓSEA; ADOLESCENTE; INFORMES DE CASOS.
ABSTRACT
Background: osteopetrosis includes a group of osseous diseases characterized by an increase of the bone density due to an osteoclastic dysfunction. It is also known as marble disease or Albers-Schönberg disease.
Objective: to present a case with marble osteopetrosis and make the review of the topic.
Clinical case: a fourteen-year-old male patient who presented bone pain that spread to the limbs, as well as severe headache that motivated a neurology study. A predominance of the left mastoid region denied the presence of articular inflammation, fever, rash, nodules or skin lesions. There were not elements of medullary hypoplasia associated to the disease of this patient. In the present physical examination no chronic disease was found. The x-rays of the patient show a widespread increase of the bone density, deformities caused by desalination and diffused bone sclerosis; all this means dense, broad bones without medullary channel which represents hypermineralized cartilages.
Conclusions: the radiologic studies were positive and confirmed the diagnosis of marble osteopetrosis (benign). The use of other imaging studies was not necessary to confirm it.
DeCS: OSTEOPETROSIS/radiography; SCLEROSIS; BONE RESORPTION; ADOLESCENT; CASE REPORTS.
INTRODUCCIÓN
El concepto de osteopetrosis (OPT) agrupa un conjunto de enfermedades óseas caracterizadas por aumento de la densidad radiográfica, debido a una disfunción osteoclástica. Esto determina una insuficiente resorción ósea, que provoca un aumento en la densidad ósea, osteoesclerosis generalizada, disminución de la resistencia ósea e incluso supresión de la médula ósea. Se la denomina también enfermedad marmórea de los huesos. 1
La osteopetrosis es una enfermedad rara en la cual la principal característica es que los huesos son demasiado densos, porque el número de osteoblastos es superior al de los osteoclastos, lo cual permite que se acumulen sales minerales. 2,3 Tiene una incidencia de 1 x 20 000 nacimientos y 1 x 200 000 adultos. En Cuba se han reportado solo 10 casos. 4,5
El primer caso de la enfermedad fue observado en 1903, por Albers-Schönberg, citado por Mazas Artasona L, 6 quien describe un paciente de 26 años, con fractura de ambos brazos por un traumatismo insignificante; radiológicamente en este paciente no se comprobó estructura ósea y la cavidad medular era inexistente y los huesos parecían de mármol.
Es una displasia hereditaria ósea causada por una disfunción, tanto del desarrollo como de la función del osteoclasto, donde se identifican mutaciones en al menos 10 genes en humanos. 7 Se puede heredar como una forma autosómica recesiva, dominante o ligada al cromosoma X, lo que constituye la forma recesiva de mayor severidad. 2,8,9
Su amplio rango de presentación varía desde un inicio neonatal con complicaciones que comprometen la vida, conocida como osteopetrosis maligna; la osteopetrosis intermedia que, en general, aparece en los 10 primeros años de vida y sus principales síntomas suelen ser más severos como: ceguera, sordera y síntomas hematológicos y la osteopetrosis benigna que se presenta en edad adulta y padecen fracturas frecuentemente y sus síntomas son: osteomielitis, dolor, artritis degenerativa y en ocasiones dolor de cabeza. 9
La gravedad de la osteopetrosis varia según la etapa de las manifestaciones clínicas: desde la forma fetal (afecta todo el esqueleto, aparece al nacimiento o durante la gestación) hasta la forma esencialmente asintomática que se diagnostica con el examen radiológico de rutina. 10
La compresión de los nervios ópticos en sus canales óseos se observa en, por lo menos, la mitad de los casos pues produce una atrofia óptica que puede presentarse desde el nacimiento; la compresión de otros pares craneanos es rara pero puede ser múltiple. 6
La disfunción osteoclástica lleva a una insuficiente resorción del cartílago calcificado e interfiere con la sustitución normal de hueso maduro, lo que provoca un aumento en la densidad ósea, osteoesclerosis generalizada, disminución de la resistencia ósea y en su forma más agresiva, supresión, por obliteración, de la actividad de la médula ósea. Se producen así huesos muy frágiles, lo que determina las fracturas múltiples, deformaciones y lesiones óseas irreversibles. Esta entidad afecta a uno de cada 200 000 niños. 11
En función de las diferencias genéticas, clínicas y evolutivas, se distinguen los siguientes tipos de osteopetrosis:
-. OPT autosómica recesiva: Dentro de las variantes recesivas se encuentra la de tipo neonatal severa o maligna, que compromete la vida, se manifiesta en los primeros meses y tiene un pronóstico no mayor de 15 años. La forma mortal: produce retraso en el crecimiento, hepatoesplenomegalia y disfunción de los nervios craneales, específicamente sordera y ceguera. En algunas ocasiones causa hidrocefalia, obliteración de la cavidad medular que produce anemia y trombocitopenia u osteoesclerosis generalizada, donde los huesos tubulares fracasan en la diferenciación cortical y medular. 12-15
-. OPT autosómica dominante: Comienza en la niñez tardía-adolescencia y se observa la típica imagen radiológica de sándwich vertebral o hueso sobre hueso (densas bandas escleróticas perpendiculares a las superficies vertebrales discales). Las principales complicaciones son óseas: fracturas, escoliosis, osteoartritis de cadera, osteomielitis, especialmente mandibular en asociación con abscesos o caries dentales. 16
-. OPT ligada al cromosoma X: con inmunodeficiencia y displasia ectodérmica y osteopatía congénita estriada que puede cursar con o sin esclerosis craneal.
El diagnóstico de OPT es esencialmente radiológico, donde se observa: aumento en la densidad ósea (densidad marmórea), osteoesclerosis intensa (imagen de hueso dentro de hueso) con pérdida de la estructura ósea, deformaciones, fusión ósea y artrosis. 17
Los cirujanos ortopédicos reportan generalmente pacientes con la variante benigna de la enfermedad (autosómica dominante), al ser las fracturas femorales, tanto del cuello como en la zona subtrocantérica, unas de las reportadas con uso de prótesis totales de cadera para el tratamiento de la coxartrosis que se produce en dicha enfermedad. 18
CASO CLÍNICO
Se presenta el caso de un paciente de 14 años de edad, color de piel blanca, masculino, sin antecedentes patológicos familiares de enfermedad reumática ni de otra enfermedad, con historia de cuadros de dolor óseo generalizado, que se iniciaba a lo largo de columna vertebral y motivaba limitaciones a las actividades diarias de juego.
Las extremidades, también fueron evaluadas por presentar cuadro doloroso de hombros que se irradiaban a lo largo de la extremidad y llegaban a ambas manos con toma de las articulaciones interfalángicas. Negó en todo momento: inflamaciones articulares periféricas, o deformidades externas visibles, fiebre, erupción, nódulos, lesiones de piel, alteraciones de los órganos de los sentidos, fracturas y traumas.
Presentó cefalea intensa asociada a todo este cuadro, lo que motivó estudio por neurología. La misma era hemicránea izquierda, con predominio de la región mastoidea de dicho lado, no se encontró características propias de la cefalea vascular, pues era permanente, no pulsátil y no tenía relación alguna su aparición con alimentos, stress o exposición a luz solar, tampoco se asoció a vértigos o zumbidos de oído.
Cabe destacar que en los episodios de reagudización de su cuadro se ha comportado con igual sintomatología, no ha tenido osteomielitis, ni fracturas, ni compromiso de los órganos de los sentidos.
En el examen físico actual no se evidencia hallazgo alguno de enfermedad inflamatoria crónica, no hepatoesplenomegalia.
El último hemograma, efectuado por consulta externa reveló un hematocrito de 0,33 %, hemoglobina de 10,8 g/l, recuento de leucocitos de 8,9 X 109 y un recuento de plaquetas de 197 X109, calcio en sangre de 2,2 mmol/l.
Las radiografías del paciente revelaron un aumento generalizado de la densidad ósea. A nivel de la columna vertebral se aprecia aumento de la densidad en las márgenes superiores e inferiores de los cuerpos vertebrales, con densidad central normal y aspecto en sandwich (figura 1).
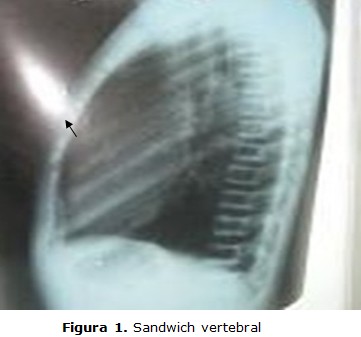
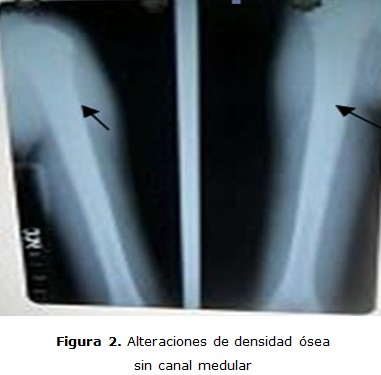
En los huesos tubulares existe hiperdensidad uniforme con desaparición del canal medular. Deformidades por desalineación de cúbito y radio, esclerosis ósea difusa, esto se traduce en huesos densos, anchos, sin canal medular, lo que representa cartílago hipermineralizado Las pruebas auditivas y visuales fueron normales (figuras 2, 3 y 4).


DISCUSIÓN
El paciente presentado tiene el antecedente de padres no consanguíneos, sumado a variados elementos clínicos y de imágenes que son compatibles con el cuadro de OPT en la modalidad de transmisión autosómica dominante.
En muchas ocasiones el diagnóstico de esta entidad es casual si no se trata de la forma maligna de la misma y se realiza por estudio de un síndrome anémico o fracturas patológicas sin explicación. 16 La anemia es el hallazgo que permite el diagnóstico en la tercera parte de los casos. A menudo se trata de una anemia hipocrómica con anisocitosis, poiquilocitosis y eritroblastosis notable; el número de leucocitos puede ser normal, raramente aumentado pero con frecuencia pueden coexistir leucopenia y trombocitopenia simulando el cuadro de una anemia aplástica. 19
Junto con la anemia es posible encontrar hepato-esplenomegalia, que inicialmente se le atribuye a una falta de desarrollo de la médula ósea pero que en realidad se debe a una hemólisis extravascular que puede mejorar con la esplenectomía, lo cual no presentó este paciente. No existió necesariamente correlación entre la severidad de la afectación ósea y la gravedad de la anemia; pues es posible encontrar pacientes con esclerosis ósea extensa sin anemia y que siguen un curso benigno. 20 No existen elementos de hipoplasia medular asociada a la enfermedad en este paciente.
Las formas malignas son causa de severas limitaciones físicas y motoras, ceguera, baja talla y hasta la muerte, provocada por infecciones, sangramientos y anemia grave. 21
Estos procesos pueden clasificarse según el predominio de la esclerosis o el defecto de modulación de los huesos en: osteoesclerosis, displasia craneotubulares e hiperostosis craneotubulares. 22
Solo uno de los padres transmitió el gen de esta enfermedad y por tanto los efectos son más leves. Pudiendo ser la enfermedad hereditaria de dos formas:
-. Cuando uno de los padres transmitió el gen de esta enfermedad y se desarrolla en una edad adulta con efectos más leves.
-. Cuando ambos son los portadores del gen y se manifiesta en el nacimiento con severos problemas los cuales llevan a la muerte.
El aumento de grosor de los huesos de la base del cráneo disminuye la luz de los agujeros de los pares craneales, lo que comprime los nervios. Esto se hace más evidente en los nervios a través de conductos más largos, como los canales ópticos, los del hipogloso o los conductos auditivos internos. Lo que trae como consecuencia que a lo largo del tiempo surjan complicaciones como ceguera y sordera. 6,22
En los pacientes con la variante recesiva y en todos con el tipo I dominante se observa dilatación de la envoltura del nervio óptico, que no fue encontrada en este paciente. La atrofia del nervio óptico y la estenosis del canal óptico se encuentran en la mayoría de los pacientes con las variantes recesiva. 22
El tratamiento en este paciente ha estado basado en el uso de analgésicos para los dolores, suplementos con vitamina D, además del uso de vitaminas y minerales que incluye el uso de sales de hierro para mantener hemoglobinas por encima de 100g/l, y el uso periódico (cada seis meses) de ozonoterapia, con el objetivo de mejorar la oxigenación de los tejidos, con lo cual ha logrado alivio de la sintomatología.
Actualmente no existe cura y en la modalidad de transmisión autosómica recesiva en su forma grave, históricamente el tratamiento ha sido paliativo. Se efectúan mayores aportes de vitamina D y parathormona, así como de fosfato, más que de calcio, para modificar el metabolismo óseo; se han usado eritropoyetina, factores estimuladores de crecimiento de colonias, incluso corticoides para estimular la hematopoyesis, con escasos resultados. En el último tiempo se ha demostrado que la terapia a largo plazo con interferón gamma 1-b aumenta la resorción ósea, la hematopoyesis y mejora la función leucocitaria. 23
Las formas malignas son causa de severas limitaciones físicas y motoras, tales como la ceguera, la baja talla y hasta la muerte, por infecciones, sangramientos y anemia grave. 24
Existen fracturas a repetición, anemia (39 % de los casos) hepatoesplenomegalia, anomalías dentarias y tendencia a osteomielitis de mandíbula. Las complicaciones neurológicas son excepcionales. 25
La supresión de la médula ósea es la complicación más severa en las formas graves o recesivas, debido a expansión ósea anormal que lleva a pancitopenia y extensión secundaria de sitios de hematopoyesis extramedular, como hígado y bazo. 26
Para iniciar un tratamiento médico es necesario detectar la enfermedad mediante una radiografía en la cual se muestra los huesos densos, además de realizar pruebas auditivas, pruebas visuales y análisis de sangre.
El diagnóstico precoz y el tratamiento oportuno evitarán las alteraciones irreversibles en la densidad ósea y el mayor riesgo de fracturas múltiples que se derivan de ello. Este evitará la obliteración del canal medular y mantendrá la función hematopoyética, así se optimiza la calidad de vida de estos pacientes y de sus familias. 28 El tratamiento curativo definitivo en el compromiso hematológico de la OPT autosómica recesiva es el trasplante de médula ósea, el cual debe realizarse de forma temprana para evitar las complicaciones. 27,29
El diagnóstico de osteopetrosis no representó mayor dificultad ni se requirieron estudios especiales de imagen (tomografía computarizada o resonancia magnética), pues bastó con el análisis radiográfico (placas simples).
CONCLUSIONES
Es indudable que este paciente presenta una forma benigna de osteopetrosis, con padres no consanguíneos, además de manifestaciones aisladas con predominio del dolor articular y la cefalea, pues los síntomas demuestran los trastornos de densidad en radiografías simples de huesos.
El diagnóstico de OPT en los pacientes asintomáticos se debe plantear cada vez que una radiografía simple evidencie elementos de displasia ósea esclerosante.
REFERENCIAS BIBLIOGRÁFICAS
1. Stark Z, Savarirayan R. Osteopetrosis. Orphanet J Rare Dis. 2009 Feb 20;4:5.
2. Cabrera Aguilar FJ, Álvarez Perea A, Gómez Antúnez M, López González-Cobos C, Pinilla Llorente B, Muiño Míguez A. Osteopetrosis del adulto. Conceptos actuales. REEMO [Internet]. 2009 [citado 3 Mar 2012];18(4):[aprox. 2 p.]. Disponible en: http://www.elsevier.es/sites/default/files/elsevier/pdf/70/70v18n04a13147273pdf001.pdf.
3. De la Uz Ruesga BO, Rodríguez Reyes I, Suárez Beyries LC, Rodríguez Brunet M, Hernández Galano G. Osteopetrosis. Medisan [Internet]. 2008 [citado 3 Mar 2012];12(1):[aprox. 3 p.]. Disponible en: http://www.bvs.sld.cu/revistas/san/vol12_1_08/san15108.pdf.
4. Al-Aama JY, Dabbagh AA, Edrees AY. A newly described mutation of the CLCN7 gene causes neuropathic autosomal recessive osteopetrosis in an Arab family. Clin Dysmorphol. 2012 Jan;21(1):1-7.
5. Raya Jiménez MA, Sequí Canet JM, Sifre Aranda M, Collar del Castillo JI. Malignant infantile osteopetrosis: usefulness of molecular diagnosis. An Pediatr. 2011 Oct;75(4):281-2.
6. Mazas Artasona L. Osteopetrosis o enfermedad de ALBERS-SCHÖNBERG. Hamburg [Internet]. 2014 [citado 3 Mar 2015];7:[aprox. 1 p.]. Disponible en: http://www.elbaulradiologico.com/
7. Villa A, Vezzoni P, Frattini A. Osteopetrosis and inmunodeficiencias in humans. Curr Opin Allergy Clin Immunol. 2006;6(6):421-7.
8. Quesada Vento HM, Fonseca Infante SM, González Rodríguez Y. Enfermedad de Albert-Schonberg: presentación de un caso. Rev Méd Electrón [Internet]. 2012 Sep-Oct [citado 3 Mar 2012];34(5):[aprox. 3 p.]. Disponible en: http://www.revmatanzas.sld.cu/revista%20medica/ano%202012/vol5%202012/tema10.htm
9. Asagiri M, Takayanagi H. The molecular understanding of osteoclast dif-ferentiation. Bone. 2007 Feb;40(2):251-264.
10. Granados Sandoval E, Martínez Estrada JG, Zepeda Cianca R, Trejo Pimentel A, Sandoval González C. Osteopetrosis (enfermedad de Albers-Schonberg): reporte de un caso y revisión clínica. Med Int Mex [Internet]. 2007 [citado 29 Abr 2012];23(6):[aprox. 4 p.]. Disponible en: http://www.medigraphic.com/pdfs/medintmex/mim-2007/mim076n.pdf
11. Bénichou O, Cleiren E, Gram J, Bollerslev J, de Vernejoul MC, Van Hul W. Mapping of Autosomal Dominant Osteopetrosis Type II (Albers-Schönberg Disease) to Chromosome 16p13.3. Am J Hum Genet. 2001;69:647-654.
12. Hashino S, Hirota G, Hasegawa M, Chiba K. Peripheral T-cell lymphoma in a patient with osteopetrosis. Ann Hematol. 2001;80(6):376-8.
13. Zetterstrom R. Osteopetrosis (marble bone disease); clinical and pathological review. Bibl Paediat [Internet]. 1958 [citado 29 Abr 2012];14(66):[aprox. 20 p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/13499353
14. Enell H, Pehrson M. Studies on osteopetrosis. Acta Paediat. 1958 May;47(3):279-287.
15. Askmyr MK, Fasth A, Richter J. Towards a better understanding and new therapeutics of osteopetrosis. British J Haematol [Internet]. 2008 [citado 29 Abr 2012];140(6):[aprox. 3 p.]. Disponible en: http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2008.06983.x/full
16. Eraso Lara I, Arenas Planelles A, D´arrigo A, Tejero Ibáñez A. La coxartrosis en los pacientes con osteopetrosis. Su tratamiento quirúrgico. Rev Española de Cirugía Osteoarticular. 2010 Oct;244(45):137-141.
17. Pangrazio A, Cassani B, Guerrini MM, Crockett JC, Marrella V, Zammataro L, et al. RANK-dependent autosomal recessive osteopetrosis: characterization of 5 new cases with novel mutations. J Bone Mineral Res [Internet]. 2011 Nov [citado 23 Jun 2012];27(2):[aprox. 5 p.]. Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/jbmr.559/full
18. Superti-Furga A, Unger S. Nosology and Classification of Genetic Skeletal Disorders: 2006 Revision. Am J Med Genet. 2007;143A:1-18.
19. Zhang J, Li Y, Hao X, Zhang Q, Yang K, Li L, et al. Recent progress in therapeutic and diagnostic applications of lanthanides. Mini Rev Med Chem. 2011 Jul;11(8):678-94.
20. Maranda B, Chabot G, Décarie JC, Pata M, Azeddine B, Moreau A, et al. Clinical and cellular manifestations of OSTM1-related infantile osteopetrosis. J Bone Miner Res. 2008 Feb;23(2):296-300.
21. Dozier TS, Duncan IM, Klein AJ, Lambert PR, Key LL. Otologic manifestations of malignant osteopetrosis. Otol Neurotol. 2005 Jul;26(4):762-6.
22. Cure JK, Key LL, Goltra DD, VanTassel P. Cranial MR imaging of osteopetrosis. Am J Neuroradiol. 2000;21(6):1110-5.
23. Satomura K, Kon M, Tokuyama R, Tomonari M, Takechi M, Yuasa T, et al. Osteopetrosis complicated by osteomyelitis of the mandible: a case report including characterization of the osteopetrotic bone. Int J Oral Maxillofac Surg. 2007 Jan;36(1):86-93.
24. Zhang J, Li Y, Hao X, Zhang Q, Yang K, Li L, et al. Recent progress in therapeutic and diagnostic applications of lanthanides. Mini Rev Med Chem. 2011 Jul;11(8):678-94.
25. Dutra F, Baroni L, Techera M. Osteopetrosis letal hereditaria. Veterinaria (Montevideo) [Internet]. 2012 [citado 9 Feb 2015];48(186):[aprox. 6 p.]. Disponible en: http://revistasmvu.com.uy/component/content/article/52-tecnicos/155-tecnico-osteopetrosis-letal-hereditaria-enfermedad-de-los-huesos-de-marmol-en-terneros-aberdeen-angus-en-uruguay.html
26. Beers MH, Berkow R, editors. Manual Merck [Internet]. España: editorial; 2010 [citado 23 Jun 2012]. Disponible en: http://www.msd.es/publicaciones/mmerck/MM_19_270.htm
27. Key LL, Rodriguez RM, Willi SM, Wright NM, Hatcher HC, Eyre DR, et al. Long-Term Treatment of Osteopetrosis with Recombinant Human Interferon Gamma. New Engl J Med. 1995 Jun 15;332:1594-9.
28. Gerritsen EJ, Vossen JM, Fasth A, Friedrich W, Morgan G, Padmos A, et al. Bone marrow transplantation for autosomal recessive osteopetrosis. A report from the Working Party on Inborn Errors of the European Bone Marrow Transplantation Group. Journal Pediatr. 1994 Dec;125(6):896-902.
Recibido: 15 de mayo de 2015
Aprobado: 17 de junio de 2015
Dra Tania Cristobo Bravo. Especialista de II Grado en Pediatría. Máster en Atención Integral al Niño. Profesor Auxiliar. Hospital Pediátrico Provincial Dr. Eduardo Agramonte Piña, Camagüey, Cuba. Email: cbtania@finlay.cmw.sld.cu

