










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Archivo Médico de Camagüey
versión On-line ISSN 1025-0255
AMC vol.20 no.5 Camagüey sep.-oct. 2016
CASOS CLÍNICOS
Hidranencefalia congénita: reporte de un adolescente en el norte de México
Congenital hydranencephaly: report of an adolescent in the north of Mexico
Dra. María de los Ángeles Barrón Muñoz I; Dra. Cecilia Hernández Reyes II; Dra. Raquel Emilia Serna Valdés II; Dr. Jorge Torres Flores II
I Instituto de Seguridad Social al Servicio de los Trabajadores del Estado. Ciudad Lerdo. Durango, México.
II Centro de Rehabilitación e Inclusión Infantil Teletón, Gómez Palacio. Durango, México.
RESUMEN
Fundamento: la hidranencefalia es la ausencia total o casi total de los hemisferios cerebrales con persistencia de líquido cefalorraquídeo, que afecta a individuos de todo el mundo sin importar su género u origen étnico. No existe un tratamiento eficaz y curativo y la gran mayoría de los pacientes mueren antes de alcanzar el tercer año de vida, aunque en algunas excepciones pueden llegar a la mayoría de edad, donde se requiere siempre del apoyo multidisciplinario.
Objetivo: presentar el caso de un varón adolescente con hidranencefalia congénita.
Caso clínico: paciente masculino de 11 años de edad ingresado en el centro a los cinco años referido como holoprosencefalia. Fue obtenido por cesárea a las 38 semanas de gestación por ruptura de membranas con somatometría normal y Apgar de cinco, requirió maniobras de reanimación neonatal avanzada. A los 30 días su perímetro cefálico aumentó a 39 cm, siguió en aumento hasta alcanzar los 54 cm a los 6 meses de edad. No presentó control cefálico ni control del tronco, tampoco fue capaz de realizar bipedestación, ni desarrolló lenguaje ni emisión de sonidos. Una tomografía cerebral realizada a su ingreso al centro reveló islotes de parénquima cerebral con meninges y línea media, lo que correspondió a una hidranencefalia. Se presentó el caso por la baja frecuencia que, los pacientes con hidranencefalia alcanzan la adolescencia.
Conclusiones: la hidranencefalia es una enfermedad que suele ser letal, y los casos que logran sobrevivir tienen secuelas neurológicas graves y discapacitantes. Aunque se conocen algunos síndromes genéticos asociados a hidranencefalia, la mayoría de los casos son esporádicos y sin manifestaciones externas al sistema nervioso central. Si bien, la esperanza de que el caso presentado muestre mayores avances en su desarrollo neurológico no es alentadora, las terapias física, ocupacional y pulmonar pueden permitir una mejor calidad de vida.
DeCS: HIDRANENCEFALIA; PARÁLISIS CEREBRAL; ESPASTICIDAD MUSCULA; NIÑO; INFORMES DE CASOS.
ABSTRACT
Background: hydranencephaly is the total or almost total absence of the cerebral hemispheres with persistent cerebrospinal fluid, which affects individuals around the world regardless of gender or ethnicity. There is no effective and curative treatment, and most patients die before reaching the third year of life, although some exceptions can come of age, always requiring multidisciplinary support.
Objective: to present the case of an adolescent male with congenital hydranencephaly.
Clinical case: a 11-year-old male from 11 who was admitted at the age of 5 referred to as holoprosencephaly. He was born by caesarean section at 38 weeks of gestation due to rupture of membranes with normal somatometry and Apgar 5, requiring advanced neonatal resuscitation maneuvers. At 30 days head circumference increased to 39 cm and was increasing, reaching 54 cm at 6 months of age. The patient has no control head and trunk, and he was not capable of bipedalism, or developed language or sound emission. A brain scan performed on admission revealed brain parenchyma islets and meninges, corresponding to hydranencephaly. The case is presented by the infrequency with which patients reach adolescence.
Conclusions: hydranencephaly is a disease that is usually fatal, and cases that survive have severe and disabling neurological sequeles. Although some genetic syndromes associated with hydranencephaly are known, most cases are usually sporadic with no other manifestations. Though the hope that the case presented shows greater progress in their neurological development is not encouraging, physical, occupational and pulmonary therapy may allow better quality of life.
DeCS: HYDRANENCEPHALY; CEREBRAL PALSY; MUSCLE SPASTICITY; CHILD; CASE REPORTS.
INTRODUCCIÓN
La hidranencefalia (HE) es una condición congénita, esporádica, con una causa no del todo comprendida la cual se presenta con una frecuencia de 1/10 000 nacidos vivos, sin existir una predisposición por género o grupo étnico. 1 El padecimiento fue descrito por Breschet en la segunda década del siglo XIX; al principio nombrado como anencefalia hidrocefálica o hidroanencefalia por Cruveilhier y por último se denominó como hidranencefalia por Kluge y Spielmayer según citan Gardea-Loera G, et al, 2 y Pavone P, et al. 3
Esta enfermedad está definida como la ausencia completa, o casi completa de los hemisferios cerebrales con la presencia de líquido cefalorraquídeo ocupando la bóveda craneal, donde se repetan los tálamos, pedúnculos cerebrales y cerebelo. 3, 4 No hay un tratamiento curativo para estos pacientes, y la muerte ocurre por lo general antes de los tres años de vida, algunos pacientes alcanzan edades mayores y requirien en todos los casos terapia física, medidas de soporte y apoyo multidisciplinario. En el reporte se presentó el caso de un adolescente con diagnóstico de HE congénita el cual tiene seis años de seguimiento y tratamiento con fisioterapia rehabilitadora.
CASO CLÍNICO
Masculino de 11 años de edad, quien fue recibido en el centro a los cinco años y dos meses de edad como holoprosencefalia, crisis convulsivas y cuadriparesia espástica, con los antecedentes de ser producto de cuarta gestación de padres no consanguíneos, madre de 37 años de edad y padre de 39 años al momento del nacimiento del propositus. Ninguno de los padres presentaba enfermedades crónico-degenerativas concomitantes. En la familia materna existía el antecedente familiar de segundo grado de consanguinidad con hidrocefalia, ano imperforado, pie equino-varo y un integrante de la familia finado en la infancia por sospecha de error innato del metabolismo, el cual no pudo ser determinado. El paciente tuvo un control prenatal normal sin eventos adversos de importancia y fue obtenido mediante cesárea a las 38 semanas de gestación por ruptura de membranas, con un peso de 2 850 gramos, talla de 52 cm y perímetro cefálico de 33 cm al nacimiento, tuvo Apgar de cinco, requirió maniobras de reanimación neonatal avanzada. Egresado a las 48 horas de vida en binomio madre-hijo sano. Inicia manifestaciones clínicas a los 30 días de edad, con aumento del perímetro cefálico a 39 cm, el cual aumento con rapidez hasta llegar a 54 cm a los seis meses de vida. No presentó control cefálico ni control de tronco, tampoco fue capaz de realizar bipedestación, ni desarrolló lenguaje ni emisión de sonidos. Mostró crisis convulsivas tipo espasmos de los tres a cinco años de edad.
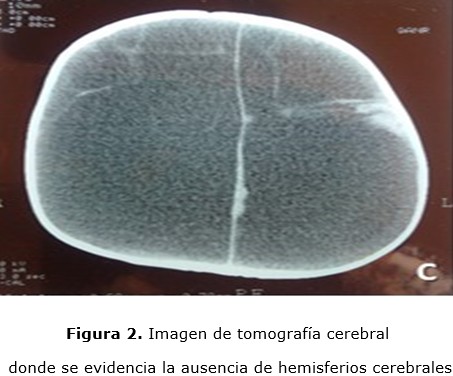
En la actualidad el paciente acude a las consultas en silla de ruedas empujada por familiares (figura 1a figura 1b), en actitud forzada por padecimiento de fondo, tiene discreto seguimiento de estímulo luminoso, presenta sonrisa social, con control de cuello regular y nulo control de tronco, con escoliosis dextroconvexa dorsolumbar; tiene tono muscular espástico grado tres en escala de Ashworth 5, 6 y grado dos en escala de Tardieu 6, 7 para miembros torácicos, y grado dos de Ashworth y uno de Tardieu para miembros pélvicos, que se traduce como nulo control volitivo en las cuatro extremidades; reflejos miotáticos exaltados, cadera izquierda luxada con Crowe tipo IV 8. Cráneo macrocéfalo con pupilas isocóricas hiporrefléxicas. Conductos auditivos externos estenóticos. Cuello simétrico con tórax normolíneo, sin anomalías cardiorrespiratorias, con abdomen globoso, depresible, no doloroso y peristalsis presente, genitales de acuerdo a edad y género. Sus valores de citología sanguínea han estado dentro de los parámetros de normalidad, así como los perfiles tiroideos, hepático y de inmunoglobulinas séricas, estás últimas realizadas por cuadros frecuentes de bronquitis y enteritis que ceden con antimicrobianos. Tiene cariotipo 46 XY con 25 metafases de lectura a una resolución de 450 bandas. Los potenciales evocados auditivos de tallo cerebral reportan un defecto severo de la conducción de la vía auditiva bilateral desde nervio periférico hasta tallo cerebral, que se traduce en hipoacusia profunda. Ultrasonografía abdominal no muestra alteraciones. La electroencefalografía revela disfunción corticosubcortical. La tomografía cerebral muestra islotes de parénquima cerebral con meninges y línea media, lo que corresponde a HE (figura 2).
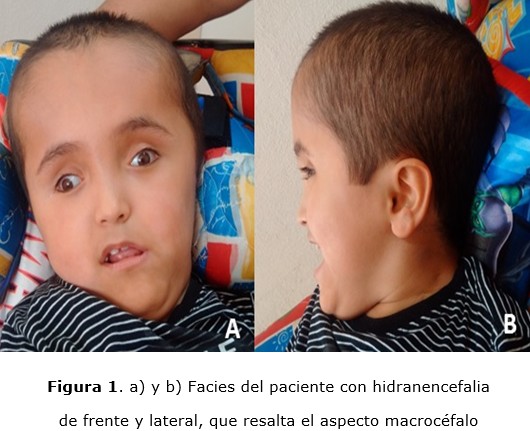
Es manejo con electroestimulación para músculos erectores de columna y para disminuir la espasticidad en bíceps braquiales, terapia ocupacional para conservar movimientos de las articulaciones de las manos, hidroterapia para neuroestimulación y disminución de tono muscular, mecanoterapia con movilización de extremidades excepto de cadera izquierda y terapia de estimulación múltiple sensorial-perceptual para sonidos, colores y texturas. Se ha manejado con diversos esquemas de aplicación de toxina botulínica tipo A diluida en solución fisiológica para músculos pectoral mayor, deltoides anterior, porción corta de bíceps braquial, palmar mayor, cubital anterior, gemelo medial y lateral, sóleo y recto anterior de cuadríceps, todos de forma bilateral.
Se definió como diagnóstico final de HE y parálisis cerebral grado V en el Sistema de Clasificación de la Función Motora Gruesa (GMFCS) para niños de 6 a 12 años. El paciente acude con regularidad al centro para continuar apoyo de fisioterapia. 8, 9
DISCUSIÓN
La HE es una condición congénita que en la mayoría de los casos suele ser letal, sin embargo, los casos que logran sobrevivir presentan complicaciones neurológicas debido al compromiso del sistema nervioso central. Si bien, se ha documentado la HE como parte importante de algunos síndromes genéticos (tabla 1).
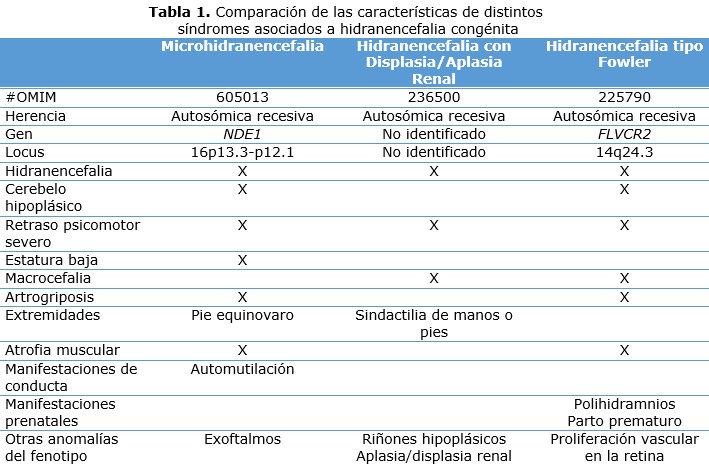
El caso que se presentó no revela datos de enfermedad concomitante; además, la mayoría de los casos de HE son atribuibles a causas ambientales, por lo general infecciosas o por posibles eventos adversos en el desarrollo vascular, como la oclusión fetal de las arterias carótidas; 4, 10 aunque no se descarta que en próximas generaciones las causas infecciosas sean mayores debido al brote de virus Zika que asola a las Américas, como evidenciaron Sarno M, et al. 11
Algo por resaltar es que la mayoría de los reportes muestran bajas expectativas de vida, con decesos antes de los tres años de edad 12, y son pocos quienes logran tener una sobrevida hasta la adolescencia, esto debido no sólo a la integridad de las estructuras que conforman el tallo cerebral, sino que también influyen los cuidados ofrecidos por la terapia física y la terapia respiratoria, que deben ser constantes durante la vida del paciente, esta forma de tratamiento aunque no está estandarizada, también la recomienda Pavone P, et al, 3 en su revisión de 2014.
A consideración de los autores, no se identifican registros latinoamericanos que muestren pacientes adolescentes con HE, y en México resalta un paciente en etapa de pubertad reportado por Gardea-Loera G y Velazco-Campos M 2 en su serie de casos.
CONCLUSIONES
Los pacientes con HE que alcanzan la adolescencia, requieren de asistencia médica especializada, para mantener en movimiento las articulaciones lo que mejora la calidad de vida.
Es probable que gracias a las técnicas de rehabilitación y a los cuidados médicos cada vez más disponibles para los pacientes con discapacidad neurológica, se observen más pacientes con HE que alcanzan la adolescencia y la edad adulta. La rehabilitación completa es difícil, debido al grave daño en el sistema nervioso central que presentan.
REFERENCIAS BIBLIOGRÁFICAS
1. Cecchetto G, Milanese L, Giordano R, Viero A, Suma V, Manara R. Looking at the missing brain: hydranencephaly case series and literature review. Pediatr Neurol. 2013 Feb;48(2):152-8.
2. Gardea-Loera G, Velazco-Campos M. Aspectos clínicos de neuroimagen y comportamiento electrofisiológico de la hidranencefalia. Arch Neurocien (Mex). 2014;19(1):48-52.
3. Pavone P, Praticó AD, Vitaliti G, Ruggieri M, Rizzo R, Parano E, et al. Hydranencephaly: cerebral spinal fluid instead of cerebral mantles. Ital J Pediatr. 2014 Oct 18;40:79.
4. Ray C, Mobley J, Thompson M, Nagy L. Hydranencephaly: Considering prolonged survival and treatment by endoscopic choroid plexus coagulation. Turk Neurosurg. 2015;25(5):788-92.
5. Ashworth B. Preliminary trial of carisoprodol in multiple sclerosis. Practitioner. 1964;192:540-2.
6. Gómez Soriano J, Cano de la Cuerda R, Muñoz Hellín E, Ortíz Gutiérrez R, Taylor JS. Valoración y cuantificación de la espasticidad: revisión de los métodos clínicos, biomecánicos y neurofisiológicos. Rev Neurol. 2012;55(4):217-26.
7. Tardieu G, Shentoub S, Delarue R. Research on a technic for measurement of spasticity. Rev Neurol (Paris). 1954;91:143-4.
8. Crowe JF, Mani VJ, Ranawat CS. Total hip replacement in congenital dislocation and dysplasia of the hip. J Bone Joint Surg. 1979;61A:15-23.
9. Palisano R, Rosembaum P, Walter S, Russell D, Wood E, Galuppi B. Development and reliability of a system to classify gross motor function in children with cerebral palsy. Dev Med Child Neurol. 1997 Apr;39(4):214-23.
10. Pachajoa H, Ariza Y, Isaza C, Méndez F. Defectos por disrupción vascular no asociados con alteraciones cromosómicas. Ginecol Obstet Mex. 2015;83:657-61.
11. Sarno M, Sacramento GA, Khouri R, Rosário MS do, Costa F, Archanjo G, et al. Zika virus Infection and stillbirths: a case of hydrops fetales, hydranencephaly and fetal demise. PLoS Negl Trop Dis. 2016;10(2):e0004517.
12. Lacunza Paredes RO, Correa López W. Hidranencefalia como presentación más severa de apoplejía cerebral fetal: a propósito de dos casos. Rev Peruana Ginecol Obstet. 2014;2:183-7.
Recibido: 8 de agosto de 2016
Aprobado: 14 de septiembre de 2016
Dr. Jorge Torres-Flores. Doctor en Genética. Centro de Rehabilitación e Inclusión Infantil Teletón, Gómez Palacio. Durango, México. Email: torresflores_md@yahoo.com.mx

