










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkVaccimonitor
versión On-line ISSN 1025-0298
Vaccimonitor v.19 n.2 Ciudad de la Habana Mayo-ago. 2010
ARTICULOS ORIGINALES
Validación de un método de determinación del polisacárido B residual en vesículas de membrana externa de Neisseria meningitidis.
Validation of a method of polysaccharide traces determination by HPLC-FL in Outer Membrane Vesicles from N. meningitidis
Jannete Rico1, Yaima Merchán2*, Matilde Cuevas2, Jenny Márquez2
1Instituto Nacional de Oncología y Radiobiología (INOR). Calle 29 y E. Vedado, Ciudad de La Habana, Cuba.
2 * Licenciada en Bioquímica, MSc en Ciencias Biológicas. Investigador agregado. Jefa del Laboratorio de Físico - Química de la Vicepresidencia de Calidad, Instituto Finlay. Instituto Finlay. Centro de Investigación - Producción de Vacunas. Ave. 27 No. 19805. AP. 16017, CP11600. La Lisa, Ciudad de la Habana, Cuba. email:ymerchan@finlay.edu.cu
RESUMEN
La Cromatografía Líquida de Alta Resolución-Fluorescencia ha resultado ser una herramienta sensible para la detección de trazas en productos farmacéuticos y es utilizada frecuentemente para la cuantificación de algunos polisacáridos de origen bacteriano que poseen ácido siálico como unidad repetitiva. En este trabajo nos planteamos como objetivo establecer y validar el método de HPLC_Fluorescencia, para determinar el contenido de polisacárido B residual en vesículas de membrana externa de Neisseria meningitidis. Para ello se realizó el montaje del método HPLC-FL, utilizando una columna de fase reversa C18 (Ultrasphere ODS, Beckman, USA) y se evaluaron seis lotes de vesículas de membrana externa de diferentes cepas Cu 385/83 y NZ 228. Se demostró que este método permitió la cuantificación de polisacárido B de una forma específica y exacta, y que cumplía con todos los parámetros de validación establecidos para un método de determinación de trazas mediante HPLC.
Palabras clave: HPLC-FL, validación, polisacárido B.
ABSTRACT
The High-Resolution Liquid Chromatography-Fluorescence has proven to be a sensitive tool for trace detection in pharmaceuticals and it is frequently used for the quantification of some bacterial polysaccharides that have sialic acid as repeating unit. In this paper we aim at establishing and validating HPLC-fluorescence to determine polysaccharide content of residual B outer membrane vesicles from Neisseria meningitidis. We assembled the HPLC-FL method to the quantification of this residue, using a C18 reverse phase column (ODS UltraSphere, Beckman, USA). We evaluated six lots of outer membrane vesicles from different strains Cu 385/83 and NZ 228. It was demonstrated that the HPLC-FL method allows the quantification of polysaccharide B in a specific and exact manner, and that it complied with the validation parameters for a method for determining trace amounts by HPLC.
Keywords: HPLC-FL, validation, polysaccharide B.
INTRODUCCION
Las vesículas de membrana externa (VME), Ingrediente Farmacéutico Activo (IFA) de las vacunas antimeningocócicas cubanas VA-MENGOC-BC® y Men B, poseen un contenido residual de polisacárido B que queda como remanente del proceso de purificación a partir de Neisseria meningitidis, serogrupo B. Para la cuantificación de la concentración de polisacárido B contaminante se utiliza el método colorimétrico, descrito por Svernnerholm, mediante el cual se cuantifican las concentraciones de polisacáridos polisialilados en muestras de proteínas semipurificadas en diferentes etapas del proceso y muestras de alto contenido polisacarídico en general (1). Sin embargo, este método no puede ser utilizado para evaluar las muestras en estudio (2) por la posible interferencia de la sacarosa, excipiente en que se encuentran conservadas.
La Cromatografía Líquida de Alta Resolución, acoplada a un detector de Fluorescencia (HPLC-Fluorescencia), ha resultado ser en nuestros días una herramienta más sensible para la detección de trazas, mediante la cual pueden ser detectados fácilmente alrededor de 0,1 pmol de polisacárido y menos de 0,2 pmoles de ácido siálico. La determinación cuantitativa del ácido N-acetilneuramínico (NANA), mediante HPLC-Fluorescencia, es utilizada frecuentemente para la cuantificación de algunos polisacáridos de origen bacteriano que poseen ácido siálico como unidad repetitiva, donde el NANA es derivatizado con O-fenilendiamina para formar un derivado de quinoxalina fluorescente estable, que es separado del exceso de reactivo mediante una columna de fase reversa C18 (3).
En la actualidad las exigencias de las empresas comercializadoras del primer mundo son mayores y requieren de un producto altamente caracterizado y controlado sobre la base de las más avanzadas técnicas de evaluación (4).
Los resultados de los procedimientos analíticos deben ser fiables, precisos y reproducibles. Los parámetros fundamentales que deben ser considerados durante la validación de los métodos de análisis para su aprobación incluyen: la exactitud, la precisión, la especificidad, la sensibilidad y la reproducibilidad (5). Una de las mayores preocupaciones de las instituciones controladoras de la calidad de los medicamentos es la confirmación de que los productos sean seguros para su comercialización. Las Buenas Prácticas de Producción constituyen un conjunto de regulaciones y requisitos que aseguran la adecuabilidad de los métodos, las instalaciones y los controles para la fabricación del medicamento, el envase y su distribución. La validación es un requisito de las Buenas Prácticas de Producción y por tanto del aseguramiento de la calidad de los productos farmacéuticos, por lo que se tiene en cuenta para realizar cualquier transacción comercial de los mismos (6).
Por estas razones se hace imperiosa la necesidad de profundizar en la caracterización de las VME y seleccionar e incorporar métodos de evaluación que refuercen los ya existentes, de manera que se incremente el aval que respalda la calidad de nuestras vacunas. Para ello nos planteamos como objetivo en este trabajo establecer y validar el método de HPLC-Fluorescencia, para determinar el contenido de polisacárido B residual de las vacunas antimeningocócicas cubanas.
MATERIALES Y METODOS
Muestras
Para realizar la validación del método fueron utilizados seis lotes de IFA, producidos en el Instituto Finlay, compuesto por VME de Neisseria meningitidis serogrupo B, conservadas en sacarosa al 3%; tres de ellos obtenidos a partir de la cepa cubana (Cu 385/83) y tres de la cepa neozelandesa (NZ 228).
Determinación de polisacárido B residual mediante HPLC-FL
Las muestras se secaron en una centrífuga de vacío a una temperatura de 60 ºC, durante 20 h. Posteriormente se realizó una hidrólisis profunda, añadiendo a los residuos secos 200 mL de ácido sulfúrico (0,1 mol/L) a 80 ºC, durante 6 h. Transcurrido este tiempo los hidrolizados se dejaron reposar durante 15 min a temperatura ambiente (25 ºC) en cámara oscura y luego se almacenaron de 2 ºC a 8 ºC hasta el día del ensayo. A cada residuo hidrolizado de las muestras y la curva se le adicionó 200 mL de una solución de OPD (orto-fenilendiamina), diluido en bisulfato de sodio (0,5 mol/L), para una concentración de 20 mg/mL. La derivatización se realizó en un horno a 80 ºC, durante 40 min.
Transcurrido este tiempo las muestras derivatizadas se dejaron reposar durante 15 min a temperatura ambiente en cámara oscura, y seguidamente se le adicionaron 600 µL de la fase móvil A (tetrahidrofurano 1%, butilamina 0,2% y ácido ortofosfórico 0,5%), se agitaron vigorosamente en vórtex y posteriormente los derivatizados se centrifugaron durante 5 min. La derivatización se realizó el mismo día del ensayo. Las muestras en todo momento se preservaron de la exposición a la luz.
Las muestras derivatizadas fueron evaluadas en un sistema HPLC-FL (Merck) utilizando un sistema precolumna-columna de fase reversa C18 (Ultrasphere ODS, Beckman; USA).
La separación se realizó a un flujo de 1 mL/min, a 40 ºC con gradiente. Los derivados fluorescentes fueron detectados a 425 nm (una longitud de onda de excitación de 230 nm);la señal cromatográfica fue registrada usando el programa Ezchrom Chromatographic data system. La determinación de la concentración de polisacáridos B se realizó utilizando una curva de calibración de ácido colomínico (polisacárido de estructura muy semejante al polisacárido B).
Validación del método analítico
Idoneidad del sistema (por tratarse de un método cromatográfico)
a) Precisión del tiempo de retención (tr): Expresada como el coeficiente de variación (CV) del tr de todas las determinaciones realizadas (5).
b) Factor de capacidad (K') (6).
- Resolución (Rs) (6).
c) Asimetría (A) (6).
d) Número de platos teóricos (N) (6).
Precisión
Repetibilidad: Se evaluó por el mismo analista seis réplicas por día de cada una de las muestras en ensayo. Se determinó el CV entre los valores de concentración de polisacárido B obtenidas para cada una de las muestras ensayadas (7).
Precisión intermedia: Se utilizaron dos lotes de IFA, procedentes de las cepas Cu 385/83 y NZ 228. Se evaluaron seis réplicas de cada una de las muestras en ensayo por dos analistas en diferentes días. Se determinó el CV entre los valores de concentración de polisacárido B para cada una de las muestras ensayadas (7) y se compararon los resultados obtenidos por ambos analistas mediante una prueba F de Fisher y una prueba t de Student.
Exactitud
La determinación de polisacárido B se realizó a dos series de seis muestras de IFA en el mismo día, procedente de las cepas Cu 385/83 y NZ 228, a las cuales se les adicionó 1 µg de ácido colomínico. Se determinó el Intervalo de Confianza de los valores de concentración hallados, así como el CV de los porcentajes de recobrado obtenidos para cada una de las muestras estudiadas.
Linealidad y rango
Se confeccionó una curva de calibración, realizando tres réplicas de cada punto. Se analizaron los coeficientes de correlación (r) y de Determinación (r2) de la curva de calibración promedio obtenida. Se determinaron los intervalos de confianza de la pendiente y del intercepto (5) y la linealidad en el rango de trabajo de la curva de calibración (8).
Especificidad
Se utilizaron dos lotes de IFA conservados en sacarosa, procedentes de las dos cepas estudiadas y una muestra de solución amortiguadora Fosfato de Sodio-Sacarosa empleada como diluente en las muestras de IFA.
Límite de cuantificación y Límite de detección
- El límite de cuantificación (LC) fue determinado utilizando los datos obtenidos en seis curvas de calibración a través de la fórmula siguiente:
LC = 10 (DE del intercepto) / promedio de la pendiente.
- El límite de detección (LD) fue calculado utilizando los datos obtenidos en seis curvas de calibración a través de la fórmula siguiente:
LD = 3,3 (DE del intercepto) / promedio de la pendiente
RESULTADOS Y DISCUSION
Al realizar la validación de este método analítico se obtuvieron los resultados siguientes:
Adecuabilidad del sistema
Este estudio corroboró que el sistema de HPLC utilizado estaba funcionando correctamente y demostró que cumple con los requisitos establecidos para un ensayo cromatográfico (9), es decir, que:
. El CV del Tiempo de retención (Tr), igual a 0,91%, expresa que el sistema es lo suficientemente preciso para garantizar los resultados de este parámetro en la validación del método.
. La columna utilizada posee un factor de capacidad (K') igual a 11,548; indicativo de que la fase estacionaria empleada es capaz de lograr una buena separación del pico de polisacárido B.
. La Resolución (Rs) del pico de polisacárido B es igual a 1,785 y no es lo suficientemente alta como para garantizar una buena separación, requisito importante para un método cromatográfico cuantitativo. Por ello se decidió usar la altura y no el área del pico para realizar todos los cálculos de concentración de este analito, de manera que puedan eliminarse los errores originados por la baja resolución del sistema.
. El pico de polisacárido B posee una buena asimetría (A = 1,43), es decir, muy cercana a 1, valor ideal, esto posibilita la exactitud de la integración del pico y su cuantificación.
. El número de platos teóricos (N) calculado para el pico de polisacárido B fue de 45.466, lo que indica que la eficiencia de la columna es lo suficientemente alta como para garantizar la calidad del ensayo. Se demostró que factores tales como: el tamaño de partículas de la columna, la temperatura, la velocidad de flujo, la viscosidad de la fase móvil y el peso molecular del analito, no estaban influyendo negativamente en el ensayo.
Se evaluó la repetebilidad del método, donde se analizó por el mismo analista durante el día seis réplicas de tres muestras de VME, obtenidas a partir de la cepa Cu 385/83 y tres de VME a partir de la cepa NZ 228 (Tabla 1).
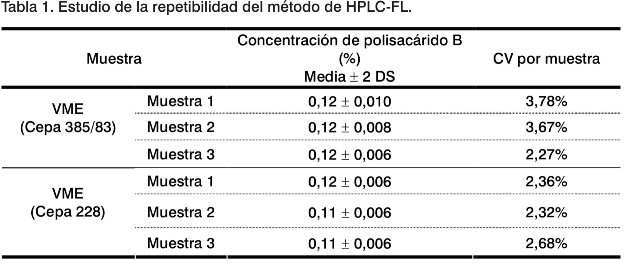
En todos los casos se obtuvo un CV menor del 5%, demostrándose que este posee una buena precisión, a pesar de ser un método cromatográfico de detección de impurezas, para los que se ha referido que coeficientes de variación menores e iguales a 10% pueden ser aceptados (10).
Precisión interensayo (precisión intermedia)
Este indicador evalúa la posibilidad del método de aportar los mismos resultados en medios diferentes, asegurando que es capaz de brindar resultados similares en momentos diferentes y en condiciones distintas, ya sean de analistas, reactivos, instrumentos, etc. (11).
En el estudio de precisión intermedia del método (Tabla 2) se observa que para todas las muestras se obtuvo un CV menor del 10%, que es aceptado teniendo en cuenta lo planteado por Rudd D (10), para un método de determinación de impurezas por HPLC, que debe ser menor e igual a 10%.
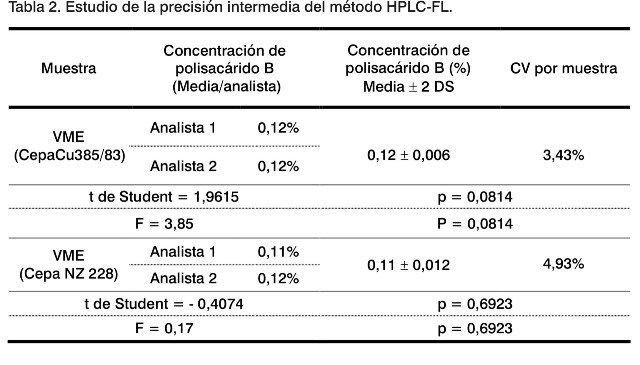
Por otra parte, se aprecia que al comparar los resultados obtenidos por los dos analistas para cada una de las muestras no existen diferencias estadísticamente significativas entre ellos (p > 0,05). Esto reafirma la precisión del método estudiado.
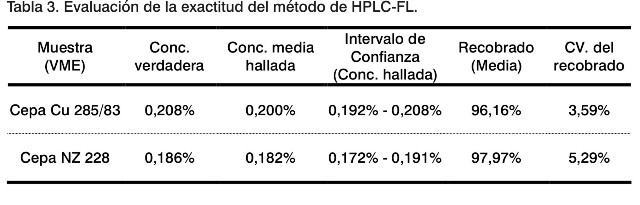
Evaluación de la exactitud
La exactitud de un método analítico depende de la matriz de la muestra, de su proceso de preparación y de la concentración del analito usada (9). El Intervalo de Confianza de los valores de concentración hallados para cada muestra incluye al valor de concentración verdadera de cada una de ellas, indicando que el método es exacto y confiable (Tabla 3). Además, todos los porcentajes de recobrados se encuentran entre 90% y 110%, con un CV < 7% para cada muestra, resultado adecuado para la determinación de impurezas (12).
Evaluación de la linealidad y el rango
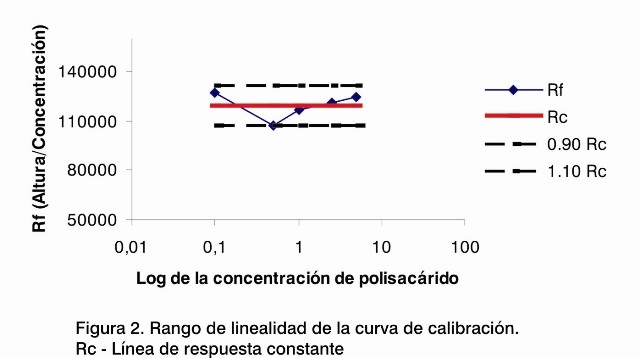
Se realizó el análisis de regresión lineal de la curva de calibración y se obtuvo un coeficiente de correlación (r) de 0,9998 y un coeficiente de determinación (r2) de 0,9997; ambos se encuentran muy cercanos a 1 y demuestran que el sistema utilizado para esta determinación tiene muy buena linealidad. Por otra parte, el CV del factor de respuesta (Rf) promedio fue de 6,53%, valor aceptado considerando que se trata de un método de determinación de trazas, donde se admite hasta un 10% de variabilidad de los resultados (Figura 1) (7).
Además, se determinó el Intervalo de Confianza para la pendiente (1,18912-1,30188) y el intercepto (-0,8164-0,11045). La pendiente es significativamente diferente de 0 (p< 0,05). Esto habla a favor de la sensibilidad del método. El intercepto incluye al cero, no existiendo diferencias estadísticamente significativas entre ellos (p > 0,05), indicativo de que el método no tiene sesgo por exceso ni por defecto.
Se muestra la relación que existe entre los factores de la respuesta para cada punto de la curva y el logaritmo de su concentración (Figura 2).
Todos los puntos se encuentran entre el 90% y el 110% de la línea de respuesta constante, evidenciando la linealidad en el rango de trabajo de la curva (8), verificándose así que este se encuentra entre 0,1 µg/mL y 5 µg/mL de concentración del polisacárido B.
Evaluación de la especificidad
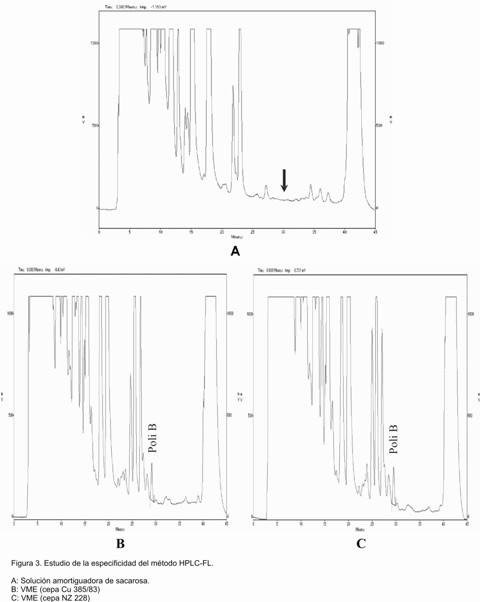
La Figura 3 muestra los cromatogramas obtenidos al estudiar la especificidad del método. En el cromatograma correspondiente a la solución amortiguadora de sacarosa (Figura 3A) no aparece ninguna otra señal en el tiempo de retención correspondiente al polisacárido B, indicativo de que ninguna otra sustancia que forma parte de las IFAs (Figuras 3 B y C), dígase excipiente, etc, eluyen en el mismo momento que el analito de interés, podemos firmar entonces que el método es específico.
Límite de cuantificación y detección
Para determinar estos parámetros se utilizaron los datos obtenidos en seis curvas de calibración.
A: Solución amortiguadora de sacarosa.
B: VME (cepa Cu 385/83)
C: VME (cepa NZ 228)
Se obtuvo un límite de cuantificación de 0,735 µg/mL que fue aceptado, teniendo en cuenta que la concentración de las muestras, evaluada mediante este método, es siempre superior a 1 µg/mL. Se obtuvo un límite de detección de 0,242 µg/mL, garantizando que el ensayo no sólo posee una buena sensibilidad para la cuantificación del polisacárido B, sino también para su detección. Esto amplía sus perspectivas de aplicación.
Teniendo en cuenta los resultados obtenidos al evaluar todos los parámetros de validación establecidos para este tipo de ensayo analítico, se puede aseverar que el método analizado se encuentra validado satisfactoriamente y constituye una herramienta valiosa para determinar los residuos de polisacárido B en Ingredientes Farmacéuticos Activos de N. meningitidis.
REFERENCIAS
1. Svennerholm L. Quantitative estimation of sialic acids II. A colorimetric resorcinol- hydrochloric acid method. Biochim Biophys Acta 1957;24:604-11.
2. Crook M. The determination of plasma or serum sialic acid. Clin Biochem 1993;26:31-8.
3. Anumula KR. Quantitative determination of monosaccharides in glycoproteins by high performance liquid cromatography with highly sensitive fluorescence detection. Anat Biochem 1994;220:277-83.
4. ICH. ICH Expert Working Group on Technical requirements for registration of pharmaceuticals for human use; Impurities in new Drug Substances (Q3A). London: ICH Press; 1999.p.1-10.
5. Centro para el Control Estatal de la Calidad de Medicamentos (CECMED). Regulación No. 41 "Validación de Métodos Analíticos ", La Habana: CECMED; 2007.
6. WHO. Technical Report Series 937. Expert Commitee on specifications for pharmaceutical preparations. Analytical method validation Geneve: WHO Press; 2006:136-140.
7. Castro M, Gascón S, Pujol M, Sans JM, Vicente L. Validación de métodos analíticos. España: Asociación Española de Farmacéuticos de la Industria;1989.
8. Huber L. Validation and Qualification in Analytical Laboratories. 2ª Edit. New York: Labcompliance; 2007.
9. FDA. Center for drug evaluation and research. Reviewer Guidance. Validation of Chromatographic methods. Editorial CDER-FDA, USA;1994.
10. Rudd DR. Suitability of Analytical methods for stability testing_Is the room for Improvement? Journal of Validation Technology 2001;5(3):255-61.
11. Fernández A, Rosales I. Validación de métodos analíticos. Segundo Taller Nacional de Validación. La Habana, Cuba: Grupo Nacional de Validación;1992.
12. Bradshaw TP. Introduction to Peptide and Protein HPLC. USA: Phenomenex;1998.
Recibido: Enero de 2010
Aceptado: Febrero de 2010


{kind=link}
{kind=link}
{kind=link}