










My SciELO
 Custom services
Custom servicesServices on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkVaccimonitor
Print version ISSN 1025-028X
Vaccimonitor vol.23 no.1 Ciudad de la Habana Jan.-Apr. 2014
ARTÍCULO ORIGINAL
Validación de un ELISA para la cuantificación de las impurezas proteicas de la cepa hospedera en el principio activo de la vacuna recombinante cubana contra la hepatitis B
Validation of an ELISA for the quantification of protein impurities from host strain on the active principle of the Cuban recombinant vaccine against hepatitis B
Eliana Pérez*, Susset Valderrama, Alain Baxera, Yunaisy Jiménez, Gerardo García, Lourdes Costa, Marisel Quintana
Centro de Ingeniería Genética y Biotecnología. Ave 31 e/ 158 y 190, Apartado Postal 6162, La Habana 10600, Cuba.
email: eliana.perez@cigb.edu.cu
* Licenciada en Química, Máster en Ciencia y Tecnología de los Procesos Biotecnológicos, Tecnólogo de Primer Nivel, Jefe del Laboratorio de Inmunoquímica de Control de la Calidad del CIGB.
RESUMEN
Se validó un inmunoensayo tipo sandwich de doble anticuerpo para cuantificar las impurezas proteicas de la cepa hospedera que pueden estar presentes en el principio activo de la vacuna cubana contra la hepatitis B. Se prepararon los reactivos biológicos empleados en el ELISA. Las proteínas de la cepa hospedera se obtuvieron bajo las mismas condiciones que el proceso de producción de la proteína recombinante hasta un paso de semipurificación o purificación primaria. Los antisueros dirigidos contra las proteínas de la cepa hospedera se generaron en conejos por un proceso de inmunización en cascada. Para la validación se analizaron los siguientes parámetros: linealidad, límite de detección y cuantificación, exactitud, rango y precisión. El ensayo establecido resultó específico, con una exactitud entre 89% y 109% para todos los tampones estudiados; se demostró un ajuste parabólico y un rango de trabajo entre 0,63 y 1,25 ng/mL, la repetibilidad y la precisión intermedia mostraron coeficientes de variación inferiores al 10 y 20% respectivamente.
Palabras clave: ELISA, hepatitis B, vacuna, validación.
ABSTRACT
A double sandwich immunoassay was validated to quantify antibody protein impurities from host strain that can be present in the active pharmaceutical ingredient of Cuban vaccine against hepatitis B. All biological reagents used in the ELISA were prepared and characterized. The proteins from the host strain were obtained under the same conditions as the production process of the recombinant protein until a purification or primary semipurification step. The antisera addressed against those proteins were generated in rabbits by an immunization process in cascade. For validation the parameters analyzed were: linearity, limit of detection and quantification, accuracy, range and precision. The established assay was specific, and the calculated accuracy was from 89% to 109% for all studied buffers. A parabolic fit and a working range from 0.63 to 1.25 ng/mL were demonstrated. The repeatability and intermediate precision showed variation coefficients below 10% and 20% respectively.
Key words: ELISA, hepatitis B, vaccine, validation.
INTRODUCCIÓN
La constante generación de nuevos productos producidos mediante la tecnología del ADN recombinante demanda que se garantice la seguridad de los mismos, por lo que las autoridades regulatorias establecen cada día estándares de calidad más elevados para su certificación y exigen que en paralelo con el desarrollo de los procesos se trabaje en el establecimiento de los métodos analíticos, los cuales deben tener una adecuada precisión y exactitud y suministrar suficientes datos para evaluar cualquier modificación en el perfil de pureza que pueda ocurrir en el producto deseado (1).
Mientras que los métodos tradicionales de análisis de la pureza de las proteínas como la electroforesis en gel de poliacrilamida en presencia de dodecilsulfato de sodio (SDS-PAGE) con tinción con plata o detección con Sypro Ruby y la electroforesis bidimensional (2D PAGE), entre otros, así como las técnicas cromatográficas, son sensibles y ampliamente aceptadas, estos métodos pueden generalmente fallar en la detección de impurezas, las cuales comigran o coeluyen con el producto.
Los procedimientos analíticos que se utilicen para la detección de las proteínas de la cepa hospedera (PCHs) deben garantizar la separación del producto deseado y las sustancias producto relacionadas de las impurezas, incluyendo productos de degradación y excipientes. Para lograr una identificación específica y una alta sensibilidad se hace necesario el empleo de métodos inmunoquímicos, como los ELISAs empleando anticuerpos policlonales (2, 3).
En este trabajo se valida un ELISA proceso específico tipo sándwich para cuantificar las proteínas de la cepa hospedera durante el proceso de purificación del ingrediente farmacéutico activo (IFA) de la vacuna recombinante cubana contra la hepatitis B (Heberbiovac HB).
MATERIALES Y MÉTODOS
Reactivos Biológicos
Las PCHs empleadas como material de referencia en el ELISA y como inmunógeno en la obtención de los anticuerpos policlonales se obtuvieron a partir de una cepa de levadura P. pastoris MP36 (his+), que no tenía insertado en su cromosoma la secuencia del gen que codifica para el antígeno de superficie de la hepatitis B (HBsAg) y se realizó el proceso específico de purificación de las impurezas proteicas bajo las mismas condiciones que el proceso de producción de la proteína recombinante, hasta un paso de semipurificación o purificación primaria (adsorción-desorción), en el cual se emplea una matriz inerte de tierra de diatomeas (HSC) (4).
Los antisueros se obtuvieron en conejo por el método de inmunización por cascada (5).
La purificación de los anticuerpos se realizó por precipitación con sulfato de amonio al 40% y posteriormente empleando una columna de inmunoafinidad que contenía gel de sepharosa CL 4B activado con BrCN (Pharmacia Fine Chemicals, Uppsala Suecia), al cual habían sido inmovilizadas las PCHs, según procedimiento establecido en nuestro laboratorio (6).
Los anticuerpos anti PCHs purificados por inmunoafinidad se conjugaron con peroxidasa de rábano picante (HRP), grado 1 (Sigma, EU) y se siguió de forma general el método de oxidación del periodato de sodio (7).
Procedimiento del ELISA proceso-específico para la cuantificación de las PCHs
Las placas de microtitulación de 96 pocillos (MaxiSorp, Nunc, EU), fueron recubiertas a la concentración previamente determinada en el ensayo de titulación, con los anticuerpos purificados por inmunoafinidad y se diluyeron en el tampón de recubrimiento (carbonato-bicarbonato de sodio 0,1 M, pH 9,6), durante 16 h a 4 °C. Se bloqueó durante 1 h a 37 °C, con 200 µL por pozo de solución bloqueadora: leche descremada (Oxoid, Inglaterra) al 0,5% en solución salina tamponada con fosfatos (SSTF) 1X (KH2PO4 0,168 mM; Na2HPO4 8,45 mM; KCl 2,61 mM y NaCl 0,137 mM, pH 7,2). Se adicionaron 100 µL de las diferentes concentraciones de la curva patrón (20; 10; 5; 2,5; 1,25 y 0,63 ng/mL) y de las muestras previamente diluidas en el tampón de ensayo (de igual composición que la solución bloqueadora). Se incubó 1 h a 37 °C. Se aplicaron 100 µL/pozo del conjugado AcPc anti PCHs-HRP en el tampón de ensayo a la dilución, previamente establecida en el ensayo de titulación. Se incubaron las placas 1 h a temperatura ambiente y en la oscuridad. Se realizaron varios lavados, se incrementaron entre los diferentes pasos a medida que transcurría el ensayo. Se añadieron 100 µL/pozo de la solución reveladora. La reacción transcurrió en la oscuridad entre 15-20 min y se detuvo con 50 µL de la solución de parada. La lectura de la absorbancia se realizó a 492 nm en un lector de microplacas (Multiskan, Flow Laboratorios). Para el procesamiento de los resultados se empleó una hoja de cálculo del Excel.
Etapas del proceso de purificación monitoreadas mediante ELISA y composición de los tampones empleados
1. Precipitación ácida (SH) (Tris 20 mM, EDTA 5 mM, KSCN 3M, NaCl 0,3 M pH 4,5).
2. Concentrado de celite (CCT) (Tris 20 mM, EDTA 3 mM, NaCl 250 mM).
3. Eluato de inmunoafinidad (EIAF) (Tris 20 mM, EDTA 3 mM, KSCN 3M, NaCl 1M).
4. Intercambio iónico positivo (IIP) (Tris 20 mM, EDTA 3 mM, NaCl 0,4 M pH 7,2).
5. Formulado final (FF) (Na2HPO4 8mM, NaH2PO4 x H2O 8 mM, NaCl 0,14 M pH 6,7).
Validación
Se realizó según las regulaciones nacionales e internacionales (8, 9). Se definieron los parámetros a estudiar según la clasificación a que pertenece el método (ensayos cuantitativos para determinar el límite de impurezas).
La linealidad se estudió al relacionar las respuestas (absorbancias), con la concentración del analito para corroborar que los datos se ajustan al modelo propuesto (regresión parabólica). Se analizaron 11 curvas preparadas de formas independientes y evaluadas en un mismo ensayo. Se calculó la ecuación y el coeficiente de determinación, el cual debe ser superior a 0,98. También se estudió la linealidad de la dilución en diferentes muestras de IFA para demostrar la condición de exceso de anticuerpo necesaria para este tipo de ensayos, para lo cual se ensayaron tres muestras a diferentes diluciones (preparadas de forma seriada) en todo el rango de concentraciones de la curva estándar.
Para el cálculo del límite de cuantificación se tomaron los resultados de 25 ensayos y se analizaron las dos concentraciones más bajas de la curva patrón (0,63 ng/mL y 1,25 ng/mL) a través de los porcentajes de recuperación entre la concentración esperada y la observada. Se determinó la exactitud y la precisión para ambas concentraciones. Para demostrar la exactitud se determinó el intervalo de confianza para los porcentajes de recuperación obtenidos. El criterio de una buena exactitud fue que el 100% de recuperación esté incluido dentro del intervalo calculado. Para demostrar la precisión se determinó el coeficiente de variación entre los porcentajes de recuperación obtenidos en los diferentes ensayos, los cuales no deben ser superiores al 20%.
La especificidad se determinó mediante el análisis del control negativo (tampón de ensayo). Se analizaron las absorbancias de diferentes tampones en los cuales se encuentran las muestras de los distintos pasos del proceso. Se realizó a través de cuatro réplicas de una misma dilución (1/100), a la cual se ensayaron los tampones y se compararon estas absorbancias con la del límite de detección del sistema. Se analizaron los límites superiores del intervalo de confianza para los tampones estudiados y el criterio de aceptación fue que el límite superior calculado para las absorbancias de cada uno de los tampones sea inferior a la absorbancia del límite de detección.
La evaluación de la exactitud se analizó mediante el comportamiento de los tampones empleados durante el proceso de purificación, por la adición de concentraciones conocidas de las PCHs (1,25 ng/mL, 3 ng/mL y 20 ng/mL) en el rango alto, medio y bajo de la curva de calibración. El ensayo se realizó por duplicado en días diferentes y se evaluó el porcentaje de recuperación para cada uno de ellos, el cual debe encontrarse entre 80-120%.
Para el establecimiento del rango de trabajo se analizaron los resultados de las dos concentraciones extremas (0,63 ng/mL y 1,25 ng/mL), para un total de 25 ensayos a través de los porcentajes de recuperación para la concentración esperada y la observada. Se determinó la exactitud y la precisión para estas concentraciones. Para demostrar la exactitud se determinó el intervalo de confianza para los porcentajes de recuperación. El criterio de una buena exactitud fue que el 100% de recuperación esté incluido dentro del intervalo calculado. Para demostrar la precisión se determinó el coeficiente de variación entre los diferentes porcentajes de recuperación, los cuales no deben ser superiores al 20%.
En el estudio de la precisión se incluyó la repetibilidad y la precisión intermedia para las muestras controles. Se analizaron en un mismo ensayo 12 réplicas de cuatro controles que se prepararon a diferentes concentraciones, de forma tal que abarcaran todo el rango de la curva estándar (bajo, medio y alto), y se determinó la media, la desviación estándar y el coeficiente de variación para cada unas de las variantes estudiadas. El criterio de aceptación es que el coeficiente de variación sea inferior al 10%. Para la precisión intermedia se estudió el comportamiento en varios ensayos, para un total de 20 réplicas correspondientes a cuatro controles de diferentes concentraciones que abarcaban todo el rango de la curva (bajo, medio y alto). Se calculó la media, la desviación estándar y el coeficiente de variación. El criterio de aceptación sería que el coeficiente de variación sea inferior al 20%.
RESULTADOS Y DISCUSION
Procedimiento del ELISA proceso-específico para la cuantificación de las PCHs
Se desarrolló un ELISA proceso-específico. Los anticuerpos obtenidos fueron usados como inmunorreactante de captura en la fase sólida y como conjugado unido covalentemente a la enzima peroxidasa. La cuantificación se realizó empleando una curva patrón de PCHs en el rango entre 20 y 1,25 ng/mL; el ensayo fue exacto, se obtuvo porcentajes de recuperación entre 80 y 120% y preciso con coeficientes de variación entre 10-20% para la repetibilidad y la precisión intermedia así como límites de detección y cuantificación de 0,63 ng/mL y 1,25 ng/mL, respectivamente, lo que posibilita su empleo en el monitoreo de los niveles de PCHs en varios pasos del desarrollo del proceso de purificación.
Validación
Como resultado de la validación del método se evidenció que la curva patrón preparada en forma de diluciones seriadas presenta linealidad en el rango entre 20 y 0,63 ng/mL, por lo que la aplicación de una función parabólica resultó adecuada para correlacionar la cantidad teórica y medida de las PCHs para cada una de las concentraciones evaluadas. Los resultados analíticos muestran una buena correlación, al obtenerse un coeficiente de determinación superior a 0,99 (R2=0,9991). Estos resultados son comparables con los reportados en la literatura (10). La condición de exceso de anticuerpos se demostró mediante el comportamiento de los lotes del IFA analizados (Fig. 1), en los cuales se observan valores constantes de concentraciones que no tienden a incrementarse con el aumento de la dilución. Este es un aspecto muy importante en un inmunoensayo para cuantificar PCHs, ya que es necesario demostrar la condición de exceso de anticuerpos como requisito para establecer la validez del ensayo (2,11), pues de esta forma se garantiza la detección de altas concentraciones de PCHs que pudieran estar presentes en una muestra. Estos resultados demuestran también la correspondencia entre las PCHs empleadas como referencia y las impurezas que pueden encontrarse en las muestras ensayadas (3).

Fig. 1. Demostración de la condición de exceso de anticuerpo. Eje de las X: 1, 2 y 3 diluciones seriadas por dos correspondientes a las muestras (3 IFAs de HBsAg hum-rec); Eje de las Y: concentración de PCHs (ng/mL).
Los resultados obtenidos en la evaluación del límite de cuantificación para las dos concentraciones más bajas de la curva patrón se muestran en la Tabla 1.

El método resultó preciso para las dos concentraciones, se obtuvo coeficientes de variación de 8,39% y 19,6% respectivamente. Sin embargo, se observa que para la concentración de 0,63 ng/mL la exactitud no fue buena ya que el 100% no estaba incluido en el intervalo calculado (75,80-89,12%). Se escogió 1,25 ng/mL como límite de cuantificación, ya que se obtuvo una media de 98% de recuperación, entre la concentración esperada y calculada, una buena precisión y exactitud, ya que el 100% estuvo incluido dentro de los límites calculados para el intervalo (desde 94,65 % hasta 101,36 %), así como un coeficiente de variación de 8,39 %. Se consideró como satisfactorio la obtención de coeficientes de variación inferiores al 20% (12). Este resultado está en correspondencia con lo esperado para este parámetro, e incluso el límite de cuantificación que se obtuvo resultó inferior a los reportados para otros ensayos de cuantificación de impurezas (12, 13), y fue similar al estuche genérico de PCHs de P. pastoris cuando se emplea el procedimiento estándar y al reportado por Dagouassat (14).
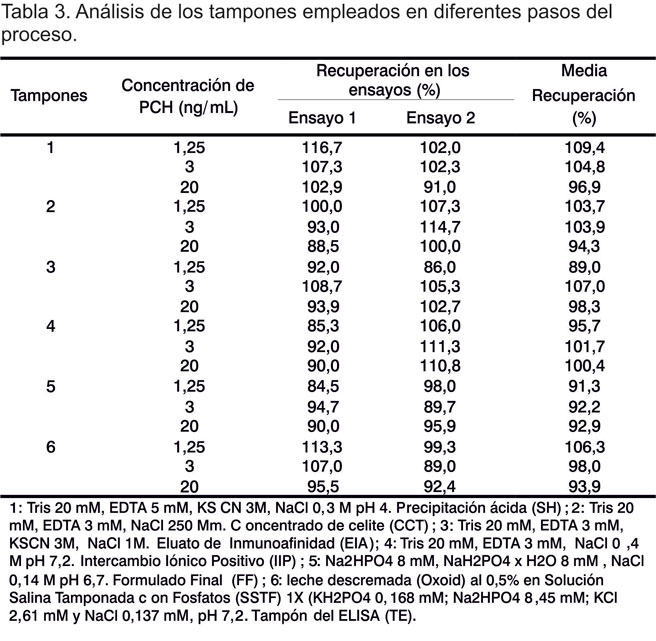
En la Tabla 2 se presentan los resultados obtenidos en la evaluación por ELISA de los diferentes tampones que intervienen en el proceso de producción.
Se aplicó una prueba de eliminación de errores groseros y se eliminó el valor de absorbancia de la réplica 1 para el EIAF. Se calculó el límite superior del intervalo de confianza para cada uno de los tampones estudiados y el resultado de todas las absorbancias fue inferior a la absorbancia del límite de detección para este ensayo (0,053). Se demostró que no hubo interferencias de los tampones evaluados. Estos resultados responden a una de las características que deben tener estos ensayos, los cuales deben ser lo suficientemente robustos como para detectar las PCHs en muestras que usualmente forman parte de matrices complejas, las cuales incluyen otras impurezas del producto como: tampones, sales y soluciones empleadas durante el desarrollo de los procesos de producción/purificación y que potencialmente pueden causar interferencias o inhibiciones (15, 16).
Se observa en la Tabla 3 el comportamiento obtenido en el ensayo de exactitud para los diferentes tampones. En este estudio se obtuvieron porcentajes de recuperación entre el 80 y el 120% para todas las concentraciones ensayadas en los diferentes tampones estudiados, los cuales se correlacionaron con los obtenidos en el estudio de diferentes matrices realizado por la Cygnus Technologies y publicado en el reporte de la validación para el estuche de P. pastoris (17). Los valores promedios de recuperación se encontraron en el rango de 89,0 y 109,4%, donde se demostró la exactitud del método. El ensayo resultó exacto para determinar las PCHs en un rango entre 20 ng/mL y 1,25 ng/mL. Esto, además, corrobora los resultados obtenidos en el estudio de la especificidad, ya que se demuestra la capacidad del método de evaluar con exactitud el analito en presencia de diferentes tampones, sin interferencias de los mismos.
El estudio de la precisión y la exactitud para las dos concentraciones extremas en curvas preparadas en diferentes días, demostró que el rango de trabajo es adecuado. Los resultados obtenidos se presentan en la Tabla 4.
Para ambas concentraciones se obtuvo una buena precisión; sin embargo, la exactitud obtenida para la concentración de 0,63 ng/mL, no fue buena, ya que el 100% no estuvo incluido en el intervalo calculado. En nuestro ensayo no consideramos la concentración de 0,63 ng/mL dentro del rango del ensayo por no ser exacta. Para garantizar una buena precisión, se realizó la cuantificación a partir de la concentración de 0,125 ng/mL, que es el límite de cuantificación calculado, ya que resultó exacto y preciso.
Los resultados obtenidos en el ensayo de exactitud también corroboran que el rango seleccionado es adecuado, ya que se obtuvieron coeficientes de variación entre 80 y 120% en la recuperación al emplear diferentes tampones. Por otra parte, se recomienda que la validación de un método de impurezas debe realizarse durante el desarrollo del propio método y el rango debe incluir el límite probable de los niveles de impurezas que deben establecerse (9). El rango calculado cumple con esta condición ya que garantiza la cuantificación de ng/mL de impurezas de PCHs, que en la evaluación de mg/mL del principio activo equivale a niveles de ppm. Estos resultados también se corresponden con los obtenidos en la literatura en el desarrollo de ensayos para detectar PCHs (10).
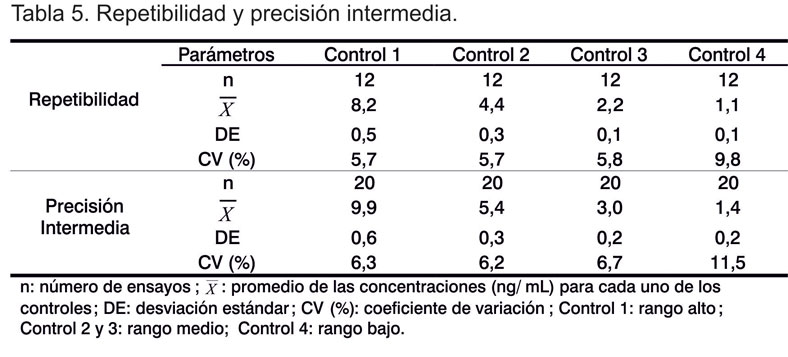
En la Tabla 5 se observan los resultados obtenidos en la evaluación de la repetibilidad y la precisión intermedia.
Para la repetibilidad se obtuvieron coeficientes de variación entre 5-6%, para muestras seleccionadas en el rango medio y alto de la curva estándar y 9,8% para el rango bajo. Para la precisión intermedia los coeficientes de variación estuvieron entre 6-7% para las muestras en el rango medio y alto y de 11,5% para el rango bajo.
Los resultados para ambos parámetros fueron inferiores al 10 y 20%, respectivamente, lo que está acorde con lo reflejado en la literatura para este tipo de ensayos (12, 13), y coincide también con varios reportes de validación publicados por la Cygnus Technologies, en el desarrollo de estuches para detectar PCHs. En ellos se considera adecuada la obtención de coeficientes de variación inferiores al 10% para los rangos medios y altos de los estudios de repetibilidad y precisión intermedia e inferiores al 12 y al 15%, respectivamente, para el rango bajo (18).
Los estudios de los diferentes parámetros de la validación resultaron similares a los obtenidos por otros autores en el desarrollo de ensayos para cuantificar PCHs, entre los que se destacan, por solo citar algunos ejemplos: un ELISA para cuantificar PCHs en el mutante SY 161 de la estafiloquinasa peguilada, capaz de detectar estas impurezas en el rango entre 1-100 ng/mL (25) y un ELISA para cuantificar proteínas de E. coli en una vacuna contra el virus sincitial humano respiratorio (hRSV), con un límite de detección de 0,5 ng/mL (14).
REFERENCIAS
1. International Conference on Harmonization (ICH). ICH Harmonized Tripartite Guideline. Comparability of Biotechnological/Biological products: Subject to Changes in Their Manufacturing Process Q5E CPMP/ICH/5721/03. Geneva: ICH; 2004.p.1-16. Disponible en: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5E/Step4 /Q5E_Guideline.pdf
2. Schwertner D, Kirchner M. Are Generic HCP Assays Outdated? BioProcess International 2010;3(1):56-61.
3. Kibbey M. Testing for Residual Host Cell Protein and DNA. Genetic Engineering and Biotechnology News 2013;33(9):1-5.
4. Centro de Ingeniería Genética y Biotecnología. Procedimiento para realizar un proceso de obtención de un material de referencia de proteínas contaminantes utilizando la cepa MP-36 de Pichia pastoris (PPO 4.26.591.99). Edición 02. La Habana: CIGB; 1999.
5. Centro de Ingeniería Genética y Biotecnología. Métodos para la obtención de sueros anti-proteínas de la cepa hospedera en conejo (PPO 4.09.150.91). Edición 07. La Habana: CIGB; 2012.
6. Centro de Ingeniería Genética y Biotecnología. Purificación de anticuerpos monoclonales anti-proteínas contaminantes de la cepa hospedera empleando columnas de inmunoafinidad (PPO 4.09.155.05). Edición 02. La Habana: CIGB; 2009.
7. Nakane PK, Kawaoi A. Peroxidase-labeled antibody. A new method of conjugation. J Histochem Cytochem 1979;22:1084-94.
8. Centro para el Control Estatal de la Calidad de los Medicamentos (CECMED). Regulación No. 37-2004: Buenas Prácticas de Laboratorio para el Control de Medicamentos. La Habana: Ediciones CECMED; 2004.
9. International Conference on Harmonization (ICH). Harmonized Tripartite Guideline. Validation of Analytical Procedures: Text and Methodology Q2 (R1). Geneva: ICH; 2005.p.1-13.
10. Wang X, Hunter AK, Mozier NM. Host Cell Proteins in Biologics Development: Identification, Quantitation and Risk Assessment. Biotechnology and Bioengineering 2009;103(3):446-58.
11. Pérez EM, Díaz N, García A, Cruz A, Martínez Y, Gil M. Montaje y Validación de un ELISA para cuantificar impurezas proteicas de Escherichia coli (E. coli) en preparaciones de Interferón Alfa 2B Humano Recombinante. Rev Farm Bioquím Univ S Paulo 1997;33(2):77-83.
12. Wang X, Schomogy T, Wells K, Mozier NM. Improved HCP Quantitation by Minimizing Antibody cross-reactivity to target proteins. BioProcess International 2010;8(1):18-24.
13. Nims R, Nixon L. Our take on process-specific vs. generic host cell protein assay. RMC Pharmaceutical Solutions Blog 2011;8:1-3.
14. Dagouassat N. Development of a quantitative assay for residual host cell proteins in a recombinant subunit vaccine against human respirator syncytial virus. Journal of Immunological Methods 2001;251(1-2):151-9.
15. Briggs J, Panfili PR. Quantitation of DNA and Protein Impurities in Biopharmaceuticals. Anal Chem 1991;63:850-9.
16. Rathore AS, Sobacke SE, Kocot TJ, Morga DR, Dufield RL, Mosier NM. Analysis for residual host cell proteins and DNA in process streams of a recombinant protein product expressed in Escherichia coli cells. J Pharm Biomed Anal 2003;32(6):1199-211.
17. Cygnus Technologies. Validation of package P. pastoris. In: Validation Summary Cygnus Technologies. Southport. Cygnus Editions; 2008. Disponible en: http//www.cygnustechnologies.com
18. Savino E, Hu B, Sellers J, Sobjak A, Majewski N, Fenton S, et al. Development of an In-House, Process-Specific ELISA for Detecting HCP in Therapeutic Antibody, Part 1. BioProcess International 2011;9(3):38-47.
Recibido: Agosto de 2013
Aceptado: Octubre de 2013


{kind=link}
{kind=link}
{kind=link}
{kind=link}