










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkVaccimonitor
versión On-line ISSN 1025-0298
Vaccimonitor vol.24 no.2 Ciudad de la Habana mayo.-ago. 2015
ARTÍCULO ORIGINAL
Implementación del algoritmo de predicción Freeman-Wimley en una aplicación web para la identificación in silico de proteínas de membrana barriles-beta
Implementation of Freeman-Wimley prediction algorithm in a web-based application for in silico identification of beta-barrel membrane proteins
José Antonio Agüero-Fernández,1* Lisandra Aguilar-Bultet,1 Yandy Abreu-Jorge,2 Agustín Lage-Castellanos,3 Yannier Estévez-Dieppa2
1 Laboratorio de Genómica Microbiana y Bioinformática. Grupo de Biología Molecular. Centro Nacional de Sanidad Agropecuaria(CENSA), Mayabeque, CP 32700, Cuba.
2 Departamento de Ciencias de la Computación. Centro Nacional de Sanidad Agropecuaria (CENSA), Mayabeque, CP 32700, Cuba.
3 Departamento de Estadística y Matemática, Centro de Neurociencias. La Habana, Cuba.
email: jaaguero@censa.edu.cu
* Licenciado en Radioquímica. Máster en Ciencias. Investigador Auxiliar. Jefe del Laboratorio de Genómica Microbiana y Bioinformática
RESUMEN
Las proteínas de tipo barril-beta desempeñan un importante papel tanto en medicina humana como veterinaria. Su localización en la superficie bacteriana y su participación en los mecanismos de virulencia de los patógenos, hacen que se hayan convertido en un interesante blanco en los estudios de búsqueda de candidatos vacunales. Freeman y Wimley desarrollaron un algoritmo de predicción basado en las propiedades físico-químicas de proteínas barriles beta transmembrana (BBTMs). Basado en el mismo y utilizando Grails, se implementó una aplicación web. Este sistema (Beta Predictor), procesa hasta 10.000 proteínas, con un tiempo de respuesta aproximado de 0,019 s por proteína de 500 residuos y permite un análisis gráfico para cada proteína. La aplicación se evaluó con un conjunto de validación de 535 proteínas no redundantes, 102 BBTMs y 433 no-BBTMs. Se calculó la sensibilidad, especificidad, coeficiente de correlación de Matthews, valor predictivo positivo y la exactitud, siendo estos 85,29%, 95,15%, 78,72%, 80,56% y 93,27%, respectivamente. El rendimiento de este sistema se comparó con el de los predictores de BBTMs, BOMP y TMBHunt y se utilizó el mismo conjunto de validación. Se obtuvieron los siguientes resultados en el orden anterior: 76,47%, 99,31%, 83,05%, 96,30% y 94,95% para el BOMP y 78,43%, 92,38%, 67,90%, 70,17% y 89,78% para el TMBHunt. El predictor BOMP superó al Beta Predictor, pero este último mostró mejor comportamiento que el TMBHunt.
Palabras clave: proteínas de membrana, barriles-beta, in silico, vacuna
ABSTRACT
Beta-barrel type proteins play an important role in both, human and veterinary medicine. In particular, their localization on the bacterial surface, and their involvement in virulence mechanisms of pathogens, have turned them into an interesting target in studies to search for vaccine candidates. Recently, Freeman and Wimley developed a prediction algorithm based on the physicochemical properties of transmembrane beta-barrels proteins (TMBBs). Based on that algorithm, and using Grails, a web-based application was implemented. This system, named Beta Predictor, is capable of processing from one protein sequence to complete predicted proteomes up to 10000 proteins with a runtime of about 0.019 seconds per 500-residue protein, and it allows graphical analyses for each protein. The application was evaluated with a validation set of 535 non-redundant proteins, 102 TMBBs and 433 non-TMBBs. The sensitivity, specificity, Matthews correlation coefficient, positive predictive value and accuracy were calculated, being 85.29%, 95.15%, 78.72%, 80.56% and 93.27%, respectively. The performance of this system was compared with TMBBs predictors, BOMP and TMBHunt, using the same validation set. Taking into account the order mentioned above, the following results were obtained: 76.47%, 99.31%, 83.05%, 96.30% and 94.95% for BOMP, and 78.43%, 92.38%, 67.90%, 70.17% and 89.78% for TMBHunt. Beta Predictor was outperformed by BOMP but the latter showed better behavior than TMBHunt.
Keywords: membrane proteins, beta-barrels, in silico, vaccine.
INTRODUCCIÓN
La caracterización de las proteínas bacterianas de superficie es un paso importante en la identificación de dianas vacunales, esto se debe a que las proteínas expuestas son la capa más externa de las bacterias y están en contacto físico directo con el hospedero y su sistema inmune. Las proteínas integrales de membrana (PIM) se dividen en dos clases estructurales, alfa-hélice (AMPs) y barriles-beta transmembrana (BBTMs).
Los BBTMs están embebidos en la membrana externa de las bacterias, mitocondrias y cloroplastos. Realizan funciones críticas y tienen un gran potencial para emplearse en vacunas o en el desarrollo de fármacos. Como ejemplos que demuestran el impacto de las proteínas barril-beta tanto en la medicina humana, como en la veterinaria, se encuentran la proteína CctA de Clostridium chauvoei (1), el antígeno protector contra la toxina del ántrax (PA) (2), la P28 y la OMP-1F de Ehrlichia chaffeensis (3), la PsrP de Streptococcus pneumoniae (4), la AatB de la Escherichia coli patógena en aves (APEC) DE205 (5) y el TPRC/D (Tp0117/131), un trímero de formación de poros de Treponema pallidum (6). Estos estudios en los que se demostró la localización en la superficie bacteriana así como su participación en los mecanismos de virulencia de los microorganismos, hacen que los BBTMs sean importantes blancos vacunales (1, 7-9).
Existen diferentes métodos para predecir las PIM. Como consecuencia de la relativa simplicidad de las estructuras AMPs, la predicción de las mismas es precisa y confiable. Sin embargo, la predicción de BBTMs representa un reto más difícil debido a su naturaleza críptica.
Existen diferentes enfoques en la predicción de la hebras trans-membrana y de la topología TM de las BBTMs. Algunos se basan en el estudio de las propiedades fisicoquímicas de las hebras β, tales como hidrofobicidad y anfipaticidad, otros en análisis estadísticos que se basan en la composición de aminoácidos de estructuras conocidas, y otros en técnicas de aprendizaje de máquina como predictores de redes neurales (NN), máquinas de soporte vectorial (SVM), modelos ocultos de Markov (HMMs), y las redes bayesianas.
Un estudio de evaluación y comparación de métodos basados en HMM que se realizó recientemente demostró que estos métodos superan a los basados en otros tipos de aprendizaje de máquina (10). La determinación de a cuál de estos programas se le puede asignar un mayor valor de confianza se obstaculiza por utilizar diferentes conjuntos de datos para la evaluación de cada uno de ellos. Recientemente, Freeman y Wimley desarrollaron un algoritmo de predicción basado en las propiedades físico-químicas de las BBTMs (11). Según los autores, la precisión es comparable a la de la predicción de AMPs.
Los objetivos de este trabajo son implementar el nuevo algoritmo desarrollado por Freeman-Wimley y crear una base de datos confiable de proteínas BBTMs y no BBTMs con el fin de comparar el desempeño de esta metodología con el BOMP y el TMB-Hunt, dos de los programas informáticos para la predicción de BBTMs, más precisos que existen.
MATERIALES Y MÉTODOS
Marco de trabajo Grails
Grails: marco de trabajo para aplicaciones web de código abierto, el cual utiliza el lenguaje de programación.
Groovy: marco de trabajo de alta productividad que sigue el paradigma de código por convención, provee un ambiente de desarrollo autónomo, oculta muchos de los detalles de configuración para el desarrollador.
Grails: se diseñó de acuerdo al patrón de arquitectura de programa MVC (modelo Vista Controlador).
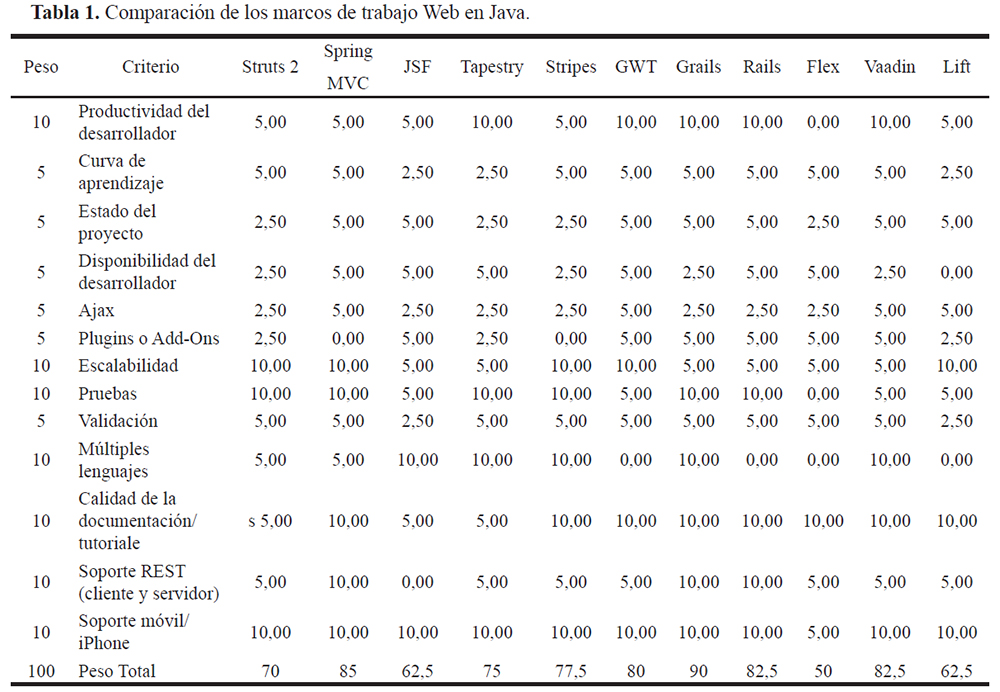
Grails: 2.2.4 se eligió después de una comparación entre diferentes marcos de trabajo y sus características (Tabla 1).
Herramientas de desarrollo
Se utilizó NetBeans 6.9 como ambiente de desarrollo integrado (IDE) de código abierto. Es una herramienta que se diseñó para que los desarrolladores, escriban, compilen, examinen y ejecuten programas. Está escrito en Java, pero se puede utilizar para programar en otros lenguajes de programación. También posee numerosos módulos y es un producto libre y sin restricciones (12).
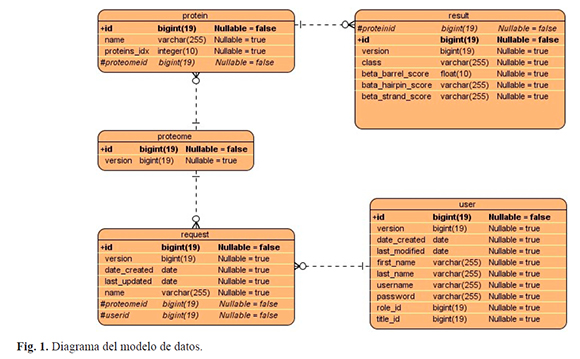
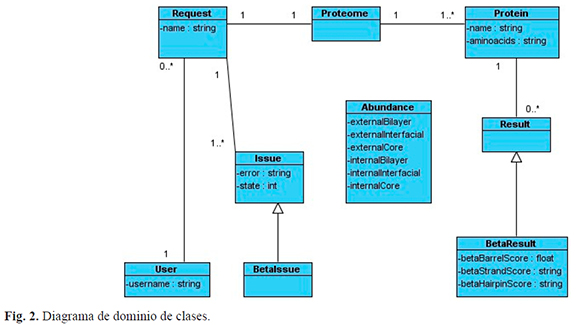
La base de datos se construyó utilizando MySQL 5.0.45 como sistema de administración de bases de datos relacionales (RDBMS) de código abierto, que provee acceso multiusuario a numerosas bases de datos. El modelo de datos está compuesto por 5 tablas (Fig. 1) que se relacionan con las clases identificadas en la lógica del negocio (Fig. 2). En la tabla user se almacenan los datos de cada usuario que complete el proceso de registro en el sistema. La identificación de datos de cada solicitud de procesamiento que realizan los usuarios se almacena en la tabla request. La información relacionada con cada proteoma, así como las proteínas que contienen los mismos, se almacenan en las tablas proteome y protein respectivamente. Cuando la solicitud se procesa, los resultados se almacenan en la tabla result.
Algoritmo
El algoritmo de predicción BBTMs que se utilizó se basó en el método de Freeman y Wimley (11).
Diagrama de clases
Se crearon nueve clases de dominio, las cuales corresponden a las principales entidades de negocio identificadas (Fig. 2). Cada clase permite el pleno acceso a la tabla de la base de datos correspondiente.
Construcción del conjunto (set) de validación
Se construyó un set de validación con proteínas cuya estructura se confirmó en el Protein Data Bank (PDB), disponible en: http://www.rcsb.org/pdb/home/home.do. Se generaron dos subconjuntos: el set de validación positivo y el negativo. Para la creación del set de validación positivo se eligieron proteínas con estructura BBTM conocida; el otro conjunto se construyó con proteínas que no poseían estructura BBTMs. El set de validación positivo se creó inicialmente con secuencias de proteínas bacterianas de 24 superfamilias de barriles- beta transmembrana a partir de la base de datos Orientations of Proteins in Membrane (OPM), la cual se encuentra disponible en: (http://opm.phar.umich.edu/). Además se incluyeron otros BBTMs diferentes con estructura resuelta. Estas últimas BBTMs surgieron como resultado de una extensa búsqueda actualizada en las bases de datos de los principales predictores BBTMs de la web.
El set de validación negativo se extrajo del PDB de proteínas transmembrana (PDBTM), disponible en:http://pdbtm.enzim.hu/, al filtrar todas las secuencias de proteínas que fueran barril-beta. Todas las secuencias de ambos conjuntos con una identidad > 50% se filtraron con la herramienta BlastClust disponible en: http://toolkit.tuebingen.mpg.de/blastclust, para evitar la existencia de secuencias redundantes. El filtrado de secuencias restantes se refinó aún más por la exclusión de las proteínas que estuvieran fuera del rango entre 60 y 4000 residuos de longitud, para cumplir con las restricciones de longitud de cadena del algoritmo de predicción de Freeman-Wimley.
Evaluación del desempeño
Para la evaluación del desempeño del Beta Predictor, se construyó una curva del tipo Receiver Operating Characteristic (ROC). Las curvas ROC son herramientas útiles para evaluar clasificadores en aplicaciones de bioinformáticas (13).
Comparación con otros predictores BBTM
Con el objetivo de seleccionar los programas con los cuales comparar el Beta Predictor para evaluar su desempeño, se realizó una búsqueda de predictores BBTMs que permitieran procesar gran cantidad de proteínas, incluso, proteomas enteros. Así el sistema implementado se comparó mediante el cálculo de algunos parámetros tales como: sensibilidad, especificidad, coeficiente de correlación de Matthews (MCC), precisión y valor predictivo positivo (VPP), con el BOMP disponible en: http://services.cbu.uib. no/tools/bomp (14) y la versión de escritorio del TMB- Hunt (15, 16).
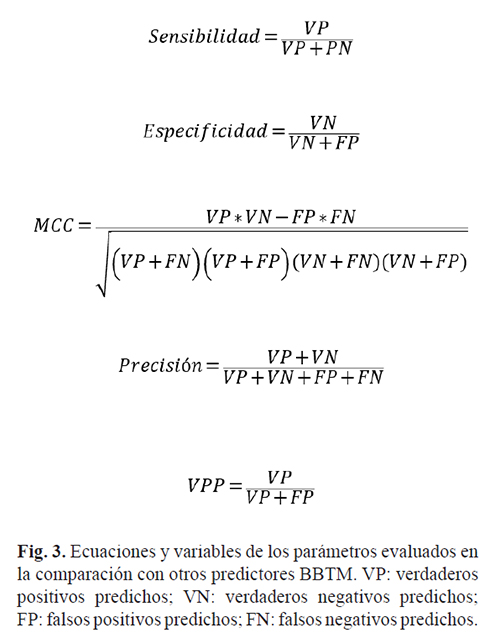
Estos parámetros se calcularon según las fórmulas que aparecen en la Figura 3.
RESULTADOS Y DISCUSIÓN
Set de validación
El set de validación quedó compuesto por 535 proteínas no redundantes, 102 conocidas como BBTMs (set de validación positivo) y 433 proteínas no BBTMs (set de validación negativo).
El set positivo se construyó principalmente con moléculas que se obtuvieron de la base de datos OMP, con el fin de garantizar la naturaleza barril-beta de la estructura de estas proteínas. OMP incluye todas las estructuras experimentales únicas. Además, el conjunto se inspeccionó de forma manual mediante una búsqueda en el PDB.
Debido a la incorporación periódica de nuevas estructuras resueltas en el PDB, este set puede mejorarse en el futuro para una mejor evaluación del rendimiento del programa.
Evaluación del desempeño
El Beta Predictor es capaz de procesar desde una secuencia de proteína hasta un proteoma predicho completo (hasta 10.000 proteínas) con un tiempo de respuesta de alrededor de 0,019 segundos por proteína de 500 residuos, permite el análisis gráfico de las predicciones para cada proteína. Además, la implementación de un proceso sincronizado permite el análisis de muchas solicitudes por usuario al mismo tiempo.
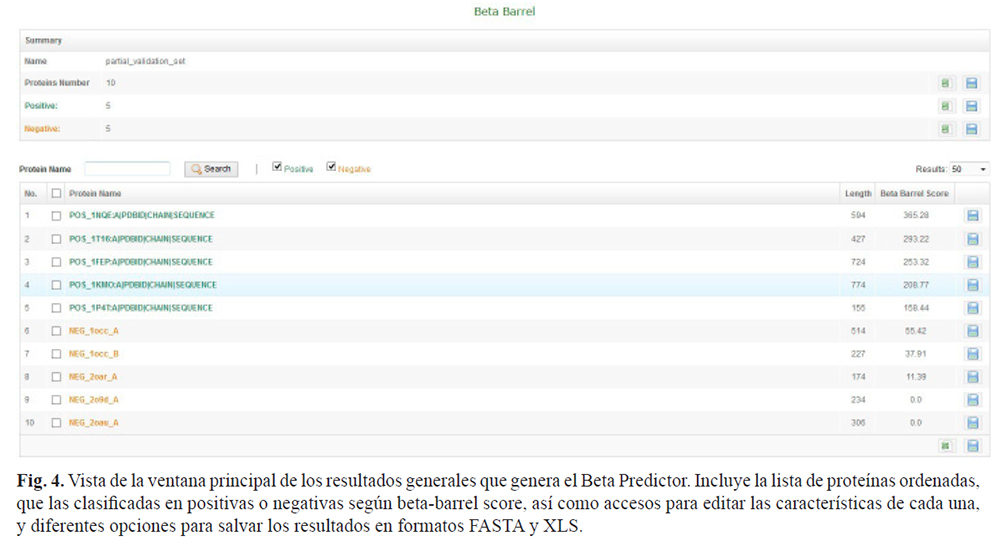
Luego de los cálculos requeridos por el algoritmo, la aplicación proporciona el acceso a los resultados globales, incluye la lista ordenada de las proteínas, clasificándolas en positivas y negativas según su grado beta barrel (beta- barrel score) (Fig. 4). La interfaz de usuario resultante permite paginar, filtrar y clasificar según los requisitos del usuario. Además, se proporciona la posibilidad de exportar los resultados a los formatos FASTA y XLS.
Adicionalmente, la solución permite el acceso a los resultados para cada proteína por separado. En todos los casos se pueden revisar los gráficos dinámicos, los residuos de interés y los valores de cadena-beta y horquilla-beta.
Los gráficos ROC muestran el rendimiento de un método de clasificación binaria con salida ordinal continua o discreta. Este tipo de gráfico muestra la sensibilidad (proporción de observaciones positivas clasificadas correctamente) y especificidad (proporción de observaciones negativas clasificadas correctamente) en la medida en que el umbral de salida se desplaza sobre todo el rango de valores posibles. Las curvas ROC no dependen de las probabilidades de clase, lo que facilita su interpretación y comparación entre diferentes conjuntos de datos (13). Originalmente se crearon para la detección de señales de radar y luego se aplicaron a la psicología y campos médicos (radiología).
En la actualidad se utilizan en la toma de decisiones médicas, bioinformática, minería de datos y aprendizaje automático, la evaluación del rendimiento de biomarcadores o la comparación de métodos de calificación. En el contexto ROC, el área bajo la curva (AUC) mide el rendimiento de un clasificador y se aplica con frecuencia para la comparación de métodos. Un valor de AUC alto significa una mejor clasificación.
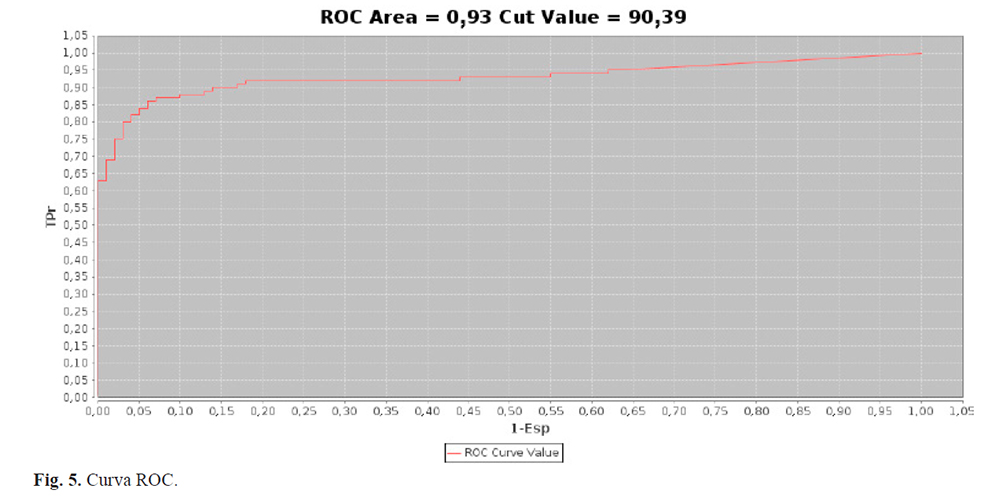
El AUC se calculó para el Beta Predictor teniendo en cuenta el set de validación construido. El valor obtenido fue de 0,93, que clasifica como muy bueno entre las categorías que se asignaron al rendimiento de los programas (13). Además, se calculó el valor de corte, obteniendo 90,39 para un 3% de falsos positivos (Fig. 5). Las proteínas cuyo beta barrel score es mayor o igual a este valor, se consideran TMMBs.
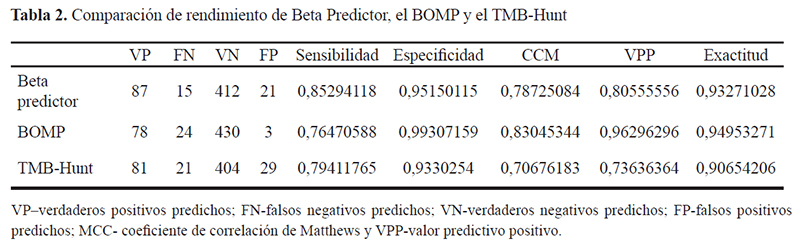
Para continuar la evaluación del rendimiento del sistema, se calcularon algunos parámetros: sensibilidad, especificidad, valor de predicción positivo (VPP), la exactitud y el coeficiente de correlación de Matthews (CCM) (Tabla 2).
Comparación con otros predictores BBTM
El rendimiento del Beta Predictor, en comparación con los predictores disponibles BOMP y TMB- Hunt, se evaluó utilizando el set de validación que se elaboró como se describió previamente (Tabla 2).
Estos programas permiten procesar proteomas enteros similar al Beta Predictor. El BOMP es un programa que combina la primera versión del algoritmo de Wimley, un reconocimiento de patrones del extremo C-terminal y un filtro final para limitar el número de falsos positivos. Se basa en el algoritmo del vecino k-más cercano (k- NN) (14). El TMB-Hunt, utiliza un algoritmo de k-NN modificado para clasificar las secuencias de proteínas como BBTM o no BBTM sobre la base de la composición de aminoácidos de toda la secuencia (15,16).
El rendimiento del Beta Predictor fue superior al del TMB-Hunt, pero inferior al del programa BOMP (con la excepción de la sensibilidad).
Esto se debió a los dos análisis adicionales que lleva a cabo el BOMP: el reconocimiento de patrones del extremo C-terminal y la eliminación de falsos positivos, el cual se realiza mediante el procedimiento de filtrado final de los valores de abundancia de aminoácidos relativos, para discriminar resultados positivos de negativos (14).
Los resultados de este estudio fueron objetivos y confiables ya que todos los programas se evaluaron mediante el mismo set de datos. El Beta Predictor puede ofrecer un mejor rendimiento entre todos los programas disponibles para la predicción de BBTMs.
La adición del reconocimiento de patrones del extremo C-terminal y el filtro para limitar el número de falsos positivos, en el programa Beta Predictor, a diferencia del BOMP, podría permitir la utilización de la última versión del algoritmo de Freeman-Wimley, mostrando resultados superiores a cualquier otro algoritmo publicado previamente para distinguir BBTMs de otras proteínas (11).
La proteínas barriles-beta tienen potencial para emplearse en vacunas o componentes de vacunas como algunas proteínas de Mannheimia haemolytica (9), la Omp87 de Pasteurella multocida serogrupo B:2 (7) y la BmaA de la cepa altamente patógénica Nagasaki (serovar 5) de Haemophilus parasuis (17).
En muchos casos, las BBTMs interactúan con las células del sistema inmune del huésped y se pueden considerar como patrones moleculares asociados a patógenos (PAMP) debido a su capacidad para señalizar por la vía de las moléculas receptoras de tipo Toll y otros receptores de reconocimiento de patrones (18). Un ejemplo de esto es la familia OmpA de Escherichia coli (8).
CONCLUSIONES
El algoritmo de Freeman-Wimley se implementó en un programa de código abierto altamente eficiente y flexible. La construcción de un set de validación de alta calidad permitió determinar, con un alto nivel de confianza, que el programa tiene un comportamiento superior a TMB- Hunt. Aunque su rendimiento fue inferior al del BOMP, si se implementan los cambios descritos previamente, el nuevo algoritmo podría transformar el sistema que se evaluó en el mejor programa de predicción de BBTMs en proteomas completos, entre los existentes y disponibles en la actualidad.
Además el Beta Predictor se puede utilizar como parte de algoritmos de predicción que tengan como objetivo la identificación de proteínas bacterianas expuestas en la superficie de estos microorganismos. Recientemente se probó con éxito en una aplicación web para la identificación in silico de posibles candidatos a vacunas bacterianas que se desarrolla en estos momentos en el Centro de Sanidad Agropecuaria (CENSA, Cuba) (19).
REFERENCIAS
1. Frey J, Johansson A, Burki S, Vilei EM, Redhead K. Cytotoxin CctA, a major virulence factor of Clostridium chauvoei conferring protective immunity against myonecrosis. Vaccine 2012;30(37):5500-5.
2. Dennis MK, Mogridge J. A protective antigen mutation increases the pH threshold of anthrax toxin receptor 2-mediated pore formation. Biochemistry 2014;53(13):2166-71.
3. Kumagai Y, Huang H, Rikihisa Y. Expression and porin activity of P28 and OMP-1F during intracellular Ehrlichia chaffeensis development. Journal of Bacteriology 2008;190(10):3597-605.
4. Schulte T, Lofling J, Mikaelsson C, Kikhney A, Hentrich K, Diamante A, et al. The basic keratin 10-binding domain of the virulence-associated pneumococcal serine-rich protein PsrP adopts a novel MSCRAMM fold. Open Biology 2014;4:130090. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/ PMC3909270/.
5. Zhuge X, Wang S, Fan H, Pan Z, Ren J, Yi L, et al. Characterization and functional analysis of AatB, a novel autotransporter adhesin and virulence factor of avian pathogenic Escherichia coli. Infection and Immunity 2013;81(7):2437-47.
6. Anand A, Luthra A, Dunham-Ems S, Caimano MJ, Karanian C, LeDoyt M, et al. TprC/D (Tp0117/131), a trimeric, pore- forming rare outer membrane protein of Treponema pallidum, has a bipartite domain structure. Journal of Bacteriology 2012;194(9):2321-33.
7. Kumar A, Yogisharadhya R, Ramakrishnan MA, Viswas KN, Shivachandra SB. Structural analysis and cross-protective efficacy of recombinant 87 kDa outer membrane protein (Omp87) of Pasteurella multocida serogroup B:2. Microbial Pathogenesis 2013;65:48-56.
8. Confer AW, Ayalew S. The OmpA family of proteins: roles in bacterial pathogenesis and immunity. Vet Microbiol 2013;163 (3-4):207-22.
9. Ayalew S, Confer AW, Hartson SD, Shrestha B. Immunoproteomic analyses of outer membrane proteins of Mannheimia haemolytica and identification of potential vaccine candidates. Proteomics 2010;10(11):2151-64.
10. Hayat S, Elofsson A. BOCTOPUS: improved topology prediction of transmembrane beta barrel proteins. Bioinformatics 2012;28(4):516-22.
11. Freeman TC, Jr Wimley WC. A highly accurate statistical approach for the prediction of transmembrane beta-barrels. Bioinformatics 2010;26(16):1965-74.
12. Brown JS, Rocher G. The Definitive Guide to Grails 2. New York: Apress; 2013.
13. Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatic 2011;12:77. Disponible en: doi:10.1186/1471-2105-12-77.
14. Berven FS, Flikka K, Jensen HB, Eidhammer I. BOMP: a program to predict integral beta-barrel outer membrane proteins encoded within genomes of Gram-negative bacteria. Nucleic Acids Res 2004;32(Suppl 2):394-9.
15. Garrow AG, Agnew A, Westhead DR. TMB-Hunt: a web server to screen sequence sets for transmembrane beta-barrel proteins. Nucleic Acids Res 2005;33(Suppl 2):188-92.
16. Garrow AG, Agnew A, Westhead DR. TMB-Hunt: an amino acid composition based method to screen proteomes for beta- barrel transmembrane proteins. BMC Bioinformatics 2005;6:56. Disponible en: doi: 10.1186/1471-2105-6-56.
17. Pina-Pedrero S, Olvera A, Perez-Simo M, Bensaid A. Genomic and antigenic characterization of monomeric autotransporters of Haemophilus parasuis: an ongoing process of reductive evolution. Microbiology 2012;158(2):436-47.
18. McClean S. Eight stranded beta -barrel and related outer membrane proteins: role in bacterial pathogenesis. Protein Pept Lett 2012;19(10):1013-25.
19. Agüero-Fernández JA, Aguilar-Bultet L, Abreu-Jorge Y, Rodríguez-Amorós Y, inventores. Predictor. Aplicación web para la identificación in silico de proteínas de superficie bacterianas. Centro Nacional de Derecho de Autor (CENDA), Cuba: Número de registro 1656-06-2014. 2014 Jun 9. Disponible en: http://baco. censa.edu.cu:8080/predictor/,http://baco.censa.edu.cu:8080/predictor/betaB/index
Recibido: Noviembre de 2014 Aceptado: Febrero de 2015


{kind=link}
{kind=link}
{kind=link}
{kind=link}