










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkVaccimonitor
versión impresa ISSN 1025-028Xversión On-line ISSN 1025-0298
Vaccimonitor vol.25 no.2 Ciudad de la Habana ago. 2016
ARTÍCULO ORIGINAL
Desarrollo de un ensayo de PCR para detectar los genes codificadores de la toxina del cólera (ctxAB) en preparaciones del candidato vacunal vivo atenuado CV638 contra el cólera
Development of a PCR assay to detect cholera toxin genes (ctxAB) in preparations of attenuated live vaccine candidate against cholera CV638
Karen Marrero-Domínguez*, Talena Ledón-Pérez y Rafael Fando-Calzada
Departamento de Biología Molecular, Área Enfermedades Infecciosas, Centro Nacional de Investigaciones Científicas, CNIC.
email: heidy.peidro@cnic.edu.cu
* Lic. Bioquímica y Biología Molecular. Dpto. Biología Molecular. Área de Enfermedades Infecciosas.
RESUMEN
El candidato vacunal vivo oral atenuado CV638 se produce siguiendo los criterios de las guías de Buenas Prácticas de Producción específicas para vacunas. El ingrediente activo de este candidato vacunal es la cepa atenuada genéticamente Vibrio cholerae 638; desarrollada por investigadores del Centro Nacional de Investigaciones Científicas de Cuba (CNIC), a partir de la cepa toxigénica de V. cholerae serogrupo O1, biotipo El Tor C7258, (Perú, 1991), mediante la remoción de los genes que codifican la producción de la toxina del cólera (ctxAB). Dado que la cepa 638 carece de estos genes en su genoma, la presencia de ctxAB en las preparaciones vacunales estaría dada por una contaminación con una cepa toxigénica de V. cholerae. El presente estudio tuvo como objetivo desarrollar un PCR específico para detectar los genes ctxAB a partir de ADN aislado de preparaciones del candidato vacunal CV638 contaminado artificialmente con V. cholerae toxigénico. La sensibilidad del ensayo de PCR empleando como molde ADN de la cepa toxigénica fue de 1 picogramo (pg) de ADN genómico por reacción, correspondiente a ~200 copias del genoma de la bacteria. La sensibilidad del método de PCR para detectar cepas toxigénicas en preparaciones vacunales de la cepa 638, contaminadas con una cepa toxigénica fue de ~7 x 103 unidades formadoras de colonia (UFC) de la cepa toxigénica por dosis del candidato vacunal CV638. Este método, una vez validado, pudiera emplearse en el control de la calidad de la producción del candidato vacunal vivo CV638.
Palabras clave: V. cholerae 638, vacunas, toxina del cólera, PCR.
ABSTRACT
The live oral vaccine candidate against cholera CV638 is produced under Good Manufacturing Practices. The active ingredient of this vaccine candidate is the genetically modified strain Vibrio cholerae 638; developed by researchers at the National Center of Scientific Research, from the strain V. cholerae serogroup O1 biotype El Tor C7258 (Peru, 1991), by removing cholera toxin genes (ctxAB). Since the strain 638 lacks these genes in its genome, the presence of ctxAB in vaccine preparations would be given by a contamination with a toxigenic V. cholerae strain. The present study aimed at designing a specific PCR to detect ctxAB genes in DNA isolated from preparations of the vaccine candidate CV638, artificially contaminated with toxigenic V. cholerae strain. Sensitivity of the optimized PCR assay was 1 pg of genomic DNA from toxigenic strain of V. cholerae C7258, corresponding to ~200 genomic copies. The sensitivity of the PCR method for detecting toxigenic strains in CV638 vaccine preparations contaminated with a toxigenic strain was ~7 x 103 colony forming units of the toxigenic strain per dose of CV638. Once validated, this method could be used in the quality control of the production of the live vaccine candidate CV638.
Keywords: V. cholerae 638, vaccines, cholera toxin, PCR.
INTRODUCCIÓN
La especie bacteriana V. cholerae enterotoxigénica de los serogrupos O1 y O139 es el agente causal del cólera (1). Aunque existen tratamientos completamente efectivos para esta enfermedad, en situaciones de brotes epidémicos muchos pacientes no reciben el tratamiento oportuno, por lo que pueden aumentar los casos de muertes. Entre el 75-85% de las infecciones ocurren en portadores asintomáticos que contribuyen además a la transmisión y a la instauración del endemismo. Por todo lo mencionado anteriormente, se han desarrollado varias vacunas contra el cólera con el objetivo de reducir el impacto en morbilidad y mortalidad de la enfermedad en esos escenarios (2). Los candidatos vacunales basados en cepas vivas atenuadas de V. cholerae se consideran una de las variantes más promisorias, dada su capacidad potencial para conferir una larga protección contra la enfermedad tras la administración de una sola dosis (3).
La cepa atenuada genéticamente V. cholerae 638 es el ingrediente activo del candidato vacunal CV638 y se obtuvo por investigadores del Centro Nacional de Investigaciones Científicas a partir de la cepa V. cholerae C7258 de serogrupo O1, biotipo El Tor y serotipo Ogawa. La cepa 638 se modificó genéticamente por deleción del profago CTXΦ, portador de los genes de la toxina del cólera (ctxAB), seguido del remplazo del gen hapA, codificante de la hemaglutinina proteasa, por un gen inactivado, mediante la inserción de un fragmento de ADN de la bacteria Clostridium thermocellum que contenía al gen celA, codificador de la endoglucanasa A (4). Esta modificación origina cepas que no expresan la hemaglutinina proteasa y poseen la actividad endoglucanasa, la cual permite distinguir a V. cholerae 638 de otros vibriones por la aparición de un halo de degradación en placas indicadoras de carboximetilcelulosa teñidas con rojo congo.
Para la liberación de los lotes vacunales de CV638 y cumpliendo las regulaciones establecidas (5) por el Centro para el Control Estatal de la Calidad de los Medicamentos (CECMED), es necesario determinar la ausencia de los genes de la toxina del cólera en las preparaciones vacunales para el control de calidad.
Aunque la cepa vacunal 638 posee el marcador celA, el cual permite su distinción fenotípica inequívoca de otras cepas de V. cholerae, la detección de pequeñas cantidades de vibriones toxigénicos mediante el análisis fenotípico de la expresión del marcador celA en presencia de hasta una cantidad 106 UFC/mL mayor de la cepa vacunal, requiere la evaluación de un gran número de colonias. Por esta razón, es necesario implementar métodos que permitan una evaluación confiable de las preparaciones vacunales y puedan ser utilizados por los laboratorios de control y producción.
La reacción en cadena de la polimerasa, conocida como PCR, en contraste con la caracterización fenotípica, permite una detección específica y sensible, y por tanto, una caracterización genotípica de las células bacterianas. Este método ha sido utilizado con éxito ampliamente para detectar cepas de V. cholerae toxigénicas presentes en muestras clínicas o ambientales (6), así como en preparados vacunales (7).
Teniendo en cuenta estos antecedentes, el presente estudio tiene como objetivo desarrollar un ensayo de PCR específico para detectar los genes de la toxina del cólera (ctxAB) y evaluar su desempeño en preparaciones del candidato vacunal vivo CV638 contaminadas artificialmente con V. cholerae toxigénico.
MATERIALES Y MÉTODOS
Cepas bacterianas, medios y condiciones de cultivo
La cepa de V. cholerae toxigénica C7258Kn (Serogrupo O1, biotipo El Tor, serotipo Ogawa, Perú, 1991), derivada de V. cholerae C7258 así como la cepa vacunal atenuada V. cholerae 638 (C7258 ΔCTXФ, hap::celA) (4) se utilizaron como fuente de ADN genómico para ser empleado como molde durante el establecimiento de las condiciones del ensayo de PCR.
Para la propagación de las cepas bacterianas se utilizó el caldo Luria-Bertani (LB) (8) y el LB sólido se preparó por adición de agar al 1,5%. Para la selección de la cepa C7258Kn el medio de cultivo se suplementó con kanamicina (Kan), a 50 μg mL-1.
Selección y síntesis de cebadores
Los cebadores se diseñaron de acuerdo con la secuencia del operón ctxAB de la cepa V. cholerae El Tor N16961 (9), mediante el análisis con el programa GeneRunner v3.05 (Hastings Software, Inc., New York, EUA). La especificidad de los cebadores se evaluó in silico mediante las herramientas bioinformáticas Primer-Blast (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) y BLASTn (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome). Una vez seleccionados los cebadores, estos se sintetizaron en el Centro de Ingeniería Genética y Biotecnología (La Habana, Cuba).
Preparación de mezclas de CV638 contaminadas con la cepa toxigénica C7258Kn
Diez dosis del candidato vacunal CV638 (1010 Unidades Formadoras de Colonias (UFC) por dosis vacunal) se resuspendieron individualmente en 1 mL de solución salina (NaCl 0,9%, m/v), se centrifugaron 10 min a 13.000 rpm y se desechó el sobrenadante. Paralelamente, a un cultivo exponencial de la cepa C7258Kn crecido en LB (Densidad óptica a 600 nm igual a 1,0) se le realizaron diluciones seriadas en base 10 hasta un factor 10-8 en solución salina. Alícuotas de 1 mL de las diluciones 10-5-10-8 de C7258Kn, por duplicado, se emplearon para resuspender los precipitados celulares de la suspensión del candidato vacunal CV638. A dos de las muestras que se emplearon como controles negativos se les añadió 1 mL de solución salina. Alícuotas de 500 μL de las suspensiones del candidato vacunal CV638 se centrifugaron 10 min a 13.000 rpm y se desechó el sobrenadante. El precipitado celular se conservó a –20ºC hasta la purificación del ADN total.
El número de UFC de la cepa C7258Kn presente en las diluciones empleadas para contaminar las preparaciones vacunales se cuantificaron por extensión en placas de LBKn, por triplicado, y en volúmenes variables según la dilución (1 mL de las diluciones 10-8 y 10-7; 100 µL de la dilución 10-6 y 100 µL de una dilución 10-1 de la dilución 10-5). El número de UFC de V. cholerae 638 presente en las preparaciones se determinó por extensión de alícuotas de 10 μL de las diluciones 10-5-10-8 en triplicado y en placas de LB. Las placas se incubaron a 37ºC durante toda la noche y al día siguiente se procedió al conteo de las colonias.
Extracción del ADN total
La purificación del ADN total bacteriano se realizó mediante el método que emplea al bromuro de cetil-trimetil amonio (CTAB) (10). El ADN genómico de la cepa C7258Kn empleado como control positivo, se aisló a partir de un cultivo en LB durante toda la noche. El ADN obtenido a partir de las preparaciones del candidato vacunal CV638 se resuspendió en 100 μL de agua destilada y se conservó a 4ºC hasta su utilización. La concentración e integridad de cada muestra se evaluó espectrofotométricamente (espectrofotómetro Eppendorf, EUA) y por electroforesis en geles de agarosa al 0,8%, respectivamente.
Establecimiento de las condiciones del ensayo de PCR
Las concentraciones de los reactivos empleadas para el ensayo de PCR fueron: MgCl2: 1,5 mM (Promega, Madison WI, U.S.A.), cebadores: 10 pmol/μL (CIGB, La Habana), deoxynucleósidos trifosfato: 200 μmol/L (Promega, Madison WI, U.S.A.), tampón de PCR 1X (Roche) y 1 U de ADN polimerasa Taq (CIGB, Sancti Spiritus). Las reacciones de PCR se realizaron en el equipo de ciclos térmicos Eppendorf (EUA) en un volumen final de 20 μL, con el siguiente programa de amplificación: desnaturalización a 94ºC por 2 min, 40 ciclos de 94ºC por 30 seg, 50ºC por 30 seg y 72ºC por 30 seg. Se utilizó un paso de extensión final de 2 min a 72ºC. Los productos amplificados se conservaron a –20ºC hasta su análisis electroforético, para lo cual se emplearon 15 μL del producto de la reacción de PCR.
Como temperatura de hibridación del ensayo se evaluaron 45, 50, 55, 60 y 65ºC y se mantuvieron constantes el resto de los parámetros de la reacción. Para seleccionar la temperatura de hibridación del ensayo, se utilizaron 100 ng de ADN molde de la cepa toxigénica C7258Kn. El programa de amplificación no sufrió variaciones excepto en el rango de temperatura de hibridación evaluado (45ºC – 65ºC).
Determinación del límite de detección del ensayo de PCR
Alícuotas de 1 µL del ADN genómico de la cepa C7258Kn (1 ng/μL-10 fg/μL) se emplearon en duplicado, en tres ensayos de PCR independientes, para determinar la sensibilidad del ensayo.
El ADN purificado a partir de cada una de las muestras del candidato vacunal 638, contaminadas con aproximadamente 10, 102, 103 y 104 células de V. cholerae C7258Kn, se empleó para determinar igualmente la sensibilidad del método. En el ensayo de PCR, se realizaron 5 réplicas por cada muestra de ADN purificada (20 en total por cada condición).
Los productos de amplificación se separaron por electroforesis en geles de agarosa al 2% (m/v) y se visualizaron por tinción con bromuro de etidio (0,5 mg/mL) e incidencia de luz ultravioleta en transiluminador (Reuser, S.L, EUA). Para el análisis densitométrico de las imágenes se empleó el programa GuefastScan (<http://www.neuronicsa.com/modulos/producto/guefast.htm>), que permite realizar la exploración automática de los carriles de un gel para determinar objetivamente la posición e intensidad relativa de las bandas de ADN, a partir del cálculo de la relación señal/ruido de la banda.
Evaluación estadística
Para todos los análisis estadísticos se empleó el paquete GraphPad Prism, versión 5 (<http://www.graphpad.com/scientific-software/prism-5-trial.software.informer.com>) y el nivel de significación se fijó en 0,05. La normalidad de los datos se verificó con la prueba de Kolmogorov-Smirnov (<http://www.physics.csbsju.edu/stats/KS-test.html>) y la homocidasticidad de múltiples grupos de muestras se comprobó por la prueba de Bartlett (<http://www.itl.nist.gov/div898/handbook/eda/section3/eda357.htm>). Se usó el análisis de varianza (ANOVA) de clasificación simple para comparar los datos de las intensidades relativas de las bandas de PCR obtenidas en las reacciones para evaluar el efecto de la temperatura de hibridación, obtenidas del análisis mediante GuefastScan. Las comparaciones a posteriori se hicieron por el Test de Tukey (<http://www.iasri.res.in/.../12-multiple comparison Procedure.pdf>).
RESULTADOS Y DISCUSIÓN
La detección de los genes de la toxina del cólera en cepas de V. cholerae aisladas de pacientes con cólera, es un paso importante en el diagnóstico de la enfermedad, debido a que solo las cepas productoras de la toxina se han asociado con epidemias y diarreas acuosas, las cuales causan deshidratación grave al paciente (11). Por este motivo, para la liberación de los lotes vacunales CV638 cumpliendo las regulaciones establecidas por el CECMED (5), es necesario determinar la ausencia de los genes ctxAB en las preparaciones de este candidato. Varios ensayos de PCR se han descrito para detectar los genes ctxAB, utilizando cebadores que amplifican regiones que abarcan tanto al gen codificador de la subunidad catalítica A (ctxA) como la subunidad estructural B (ctxB) (6, 12-14).
Selección de cebadores específicos para los genes ctxAB
La sensibilidad y especificidad de una reacción de PCR depende en buena medida de usar cebadores de calidad, que incluye que sean específicos para cada alelo del gen diana (15-18). Además, los cebadores no deben formar estructuras secundarias, tales como los lazos o burbujas, ni dímeros intermoleculares. En este estudio, se analizaron combinaciones de oligonucleótidos presentes en la región codificadora de los genes ctxAB a partir de la secuencia del genoma de V. cholerae N16961, con el objetivo de determinar la presencia de ambos genes simultáneamente. La especificidad de los cebadores se evaluó in silico, mediante la búsqueda de secuencias homólogas en las base de datos nucleotídica no redundante del NCBI. Al concluir estos análisis, se seleccionaron los oligonucleótidos de secuencias: 5´-CCC AAA GTC TAG GTG TAA AAT TCC-3´ y 5´- GCA ATC CTC AGG GTA TCC TTC-3´, los que se corresponden con los nucleótidos 686-709 de ctxA y 267-287 de ctxB, respectivamente.
Posteriormente, se realizó una evaluación de los cebadores en V. cholerae y E. coli mediante el programa Primer-Blast. Este análisis reveló que los cebadores amplifican una única banda de 399 pb, solamente en cepas de referencia de V. cholerae y no en E. coli.
Las cepas de V. cholerae identificadas pertenecían al serogrupo O1 y los biotipos El Tor (N16961, MAK757, 2010EL-1786, IEC224 y G4222) y Clásico (O395), así como al serogrupo O139 (MO10). Este análisis indicó que el par de cebadores seleccionados permiten detectar la presencia de los genes ctxAB en una gran variedad de cepas de V. cholerae, pertenecientes tanto al biotipo El Tor como Clásico, del serogrupo O1 así como el serogrupo O139.
La calidad de los cebadores informados en la literatura que han sido utilizados para detectar los genes de patogenicidad de V. cholerae, incluidos los empleados para detectar ctxA y ctxB, se analizó in silico recientemente (19-20). Este análisis reveló que dos terceras partes de los cebadores publicados no son capaces de detectar apropiadamente todas las variantes genéticas de los genes diana.
En el caso de ctxA, de acuerdo al análisis del alineamiento de 10 secuencias únicas derivadas de 65 secuencias informadas, los 18 cebadores analizados fueron capaces de identificar todos los alelos de ctxA secuenciados (19-20). En el caso del cebador seleccionado en el presente estudio, que hibrida con el gen ctxA, el alineamiento de las secuencias correspondientes a 9 secuencias únicas de ctxA reveló que este cebador hibrida en las regiones conservadas en todas las secuencias, excepto en la del fragmento ctxAB de una cepa de V. cholerae serotipo Inaba aislada en Vietnam (número de acceso AJ575590.1) (21).
En el caso del gen ctxB, de acuerdo al análisis del alineamiento de 24 secuencias únicas derivadas de 205 secuencias informadas, ninguno de los cebadores publicados es capaz de identificar a todos los alelos de ctxB secuenciados (19-20). En el caso del cebador seleccionado que hibrida con el gen ctxB, este no se encuentra en la región conservada en todas las secuencias de ctxB. Sin embargo, hasta el momento, entre las cepas de V. cholerae de diferentes serogrupos y biotipos se han descrito 9 genotipos diferentes de ctxB, sobre la base de variaciones conservadas no aleatorias en los nucleótidos 58, 72, 83, 101, 115, 138, 165 y 203 (21). El análisis de estas variaciones reveló que el cebador en ctxB no hibrida en estas secuencias (nucleótidos 267-287), por lo que debe ser capaz de reconocer todos los genotipos descritos.
La especificidad de los cebadores se evaluó experimentalmente utilizando como ADN molde el de las cepas V. cholerae C7258Kn y V. cholerae 638. El par de cebadores amplificó un único fragmento de ADN en la cepa toxigénica, no así en la vacunal que tiene delecionados los genes ctxAB (Fig. 1, carriles 1-2).
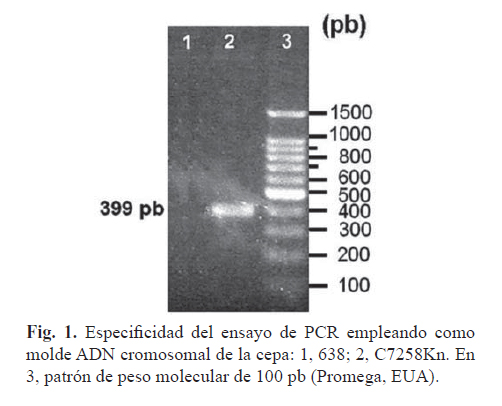
El tamaño del fragmento amplificado fue de 400 pb, según el análisis de su migración y su comparación con la del patrón de peso molecular. Esta talla es cercana a la esperada (399 pb). Estos resultados indicaron que los cebadores seleccionados eran específicos para los genes ctxAB.
Rango de temperatura de hibridación útil para el par de cebadores seleccionados
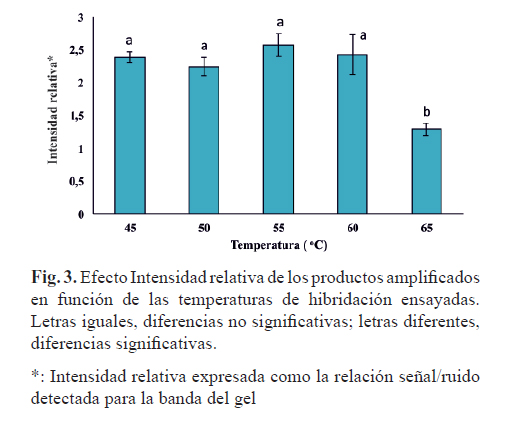
Teniendo en cuenta la temperatura de fusión de los cebadores, predicha por el programa GeneRunner (60,1ºC para el cebador 1 y 61,2ºC para el cebador 2), se evaluaron como temperaturas de hibridación 45, 50, 55, 60 y 65ºC. El par de cebadores amplificó el fragmento esperado a todas las temperaturas evaluadas (Fig. 2, carriles 1-5).

La intensidad relativa de los fragmentos amplificados se determinó mediante el análisis densitométrico de la imagen de los geles con el programa GueFastScan (Fig. 3). En el gráfico se muestran el promedio de la relación señal/ruido de cada banda y la desviación estándar de 3 réplicas de un mismo experimento por duplicado. La comparación de la intensidad relativa del producto de amplificación con los cebadores a las diferentes temperaturas de hibridación, mediante ANOVA de clasificación simple (α=0,05; g.l.e.=29), reveló que no existen diferencias significativas en la intensidad relativa del producto amplificado a las temperaturas de hibridación de 45, 50, 55 y 60ºC y sí existen diferencias significativas a 65ºC, según análisis con Prueba de Tukey. Como temperatura de hibridación se seleccionó 50ºC, dado que se observó una amplificación específica de la banda esperada.
Límite de detección del ensayo de PCR
La sensibilidad del ensayo de PCR frente al ADN de la cepa toxigénica C7258Kn se evaluó preliminarmente utilizando cantidades decrecientes de ADN (1 ng hasta 10 fg por reacción) por triplicado. Las reacciones que amplificaron consistentemente la banda esperada fueron aquellas en las que se encontraban como mínimo 1 pg de ADN molde, valor estimado como límite de detección de este ensayo (Fig. 4). Teniendo en cuenta que el genoma de V. cholerae posee 3,2 Mb, 1 pg de ADN se corresponden con aproximadamente 200 genomas de la bacteria. Dado que los genes ctxAB se encuentran en simple copia, mediante este ensayo es posible detectar hasta ~200 copias de los genes ctxAB por reacción.

Aunque la amplificación a partir de ADN puro de la cepa toxigénica es relativamente simple, la verdadera prueba de los métodos basados en un PCR para detectar cepas toxigénicas de V. cholerae en una preparación de una cepa vacunal viva, es la detección de su sensibilidad en condiciones donde exista un alto número de células de la cepa vacunal, así como lio-protectores tales como leche y sacarosa que pueden interferir durante la purificación del ADN. Por esta razón, el ADN purificado a partir las dosis de CV638 contaminadas con las diferentes cantidades de la cepa toxigénica (Tabla 1) se empleó como molde en el ensayo de PCR para determinar el límite de detección en estas condiciones, empleando 2 µg de ADN total por reacción. Se observó una correspondencia entre el número de UFC de la cepa toxigénica en las dosis del CV638 contaminadas con las diluciones realizadas (Tabla 1). El número de UFC de 638 en la preparación vacunal fue de 9,8 ± 1,5 x 1010.
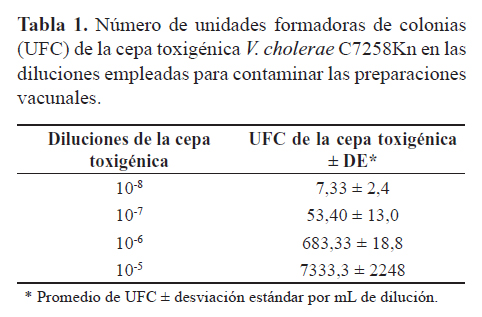
Este ensayo amplificó la banda de la talla esperada (399 pb) en las muestras de ADN provenientes de las preparaciones contaminadas con ~7 x 103 células de la cepa toxigénica V. cholerae C7258Kn (Fig. 5). Este valor está en el mismo orden que el reportado por otros autores para garantizar la seguridad de vacunas orales vivas contra el cólera como es el caso de la vacuna Orochol (7) por lo que, una vez validado, el ensayo del presente estudio pudiera emplearse en la detección de cepas toxigénicas en los preparados vacunales. Otros métodos de PCR tales como el PCR anidado y el PCR en tiempo real, pudieran mostrar una sensibilidad mayor; pero desde el punto de vista técnico son más exigentes.
La dosis infectiva de V. cholerae O1 se ha estimado sea de 105–108 UFC totales en individuos sanos, aunque en algunos individuos con baja acidez estomacal se ha estimado que pudiera llegar a ser de hasta 10³ UFC/mL (1). La comparación de estas dosis infectivas con el nivel de detección de ~7 x 103 células de la cepa toxigénica por preparación vacunal, indica que el nivel de detección del ensayo descrito es adecuado para ser empleado durante el control de calidad de los lotes vacunales y pudiera, por tanto, ser una herramienta valiosa en el aseguramiento de la calidad de los lotes del candidato vacunal vivo CV638. Para ello será necesario validar la técnica de acuerdo a las regulaciones establecidas por el CECMED (5) entre las que se encuentran determinar el límite de detección, la especificidad y la robustez del método. En este caso, se determinó el límite de detección y la sensibilidad del método, dos características importantes del ensayo.
REFERENCIAS
1. Harris JB, La Rocque RC, Qadri F, Ryan ET, Calderwood SB. Cholera. Lancet 2012;379(9835):2466-76.
2. O’Ryan M, Vidal R, Del Canto F, Carlos SJ, Montero D. Vaccines for viral and bacterial pathogens causing acute gastroenteritis: Part I: Overview, vaccines for enteric viruses and Vibrio cholerae. Hum Vaccin Immunother 2015;11(3):584-600.
3. Pastor M, Pedraz JL, Esquisabel A. The state-of-the-art of approved and under-development cholera vaccines. Vaccine 2013;31(38):4069-78.
4. Benítez JA, García L, Silva A, García H, Fando R, Cedré B, et al. Preliminary assessment of the safety and immunogenicity of a new CTXPhi-negative, hemagglutinin/protease-defective El Tor strain as a cholera vaccine candidate. Infect Immun 1999;67(2):539-45.
5. Centro para el control estatal de la calidad de los medicamentos (CECMED). Validación de métodos analíticos. Regulación No. 41-2007. La Habana: CECMED; 2007.
6. Dalusi L, Lyimo TJ, Lugomela C, Hosea KM, Sjoling S. Toxigenic Vibrio cholerae identified in estuaries of Tanzania using PCR techniques. FEMS Microbiol Lett 2015;362(5): doi:10.1093/femsle/fnv009.
7. Studer E, Candrian U. Development and validation of a detection system for wild-type Vibrio cholerae in genetically modified cholera vaccine. Biologicals 2000;28(3):149-54.
8. Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A Laboratory Manual. 2nd ed. New York: Cold Spring Harbor; 1989.
9. Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dudson RJ, et al. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 2000;406(6795):477-83.
10. Ausubel FM, Brent R, Kingston RE, Moore DD; Seidman JG, Smith JA, et al, editors. Short protocols in molecular biology. 3rd ed. New York: John Wiley & Sons; 1995.
11. WHO. Cholera, 2013. Wkly Epidemiol Rec 2014;89(31):345-56.
12. Li F, Kan B, Wang D. Development of both multiple PCR and real-time SYBR green PCR for the detection of Vibrio cholerae non-O1/O139 serogroups. Zhonghua Liu Xing Bing Xue Za Zhi. 2014;35(1):66-70.
13. Vinothkumar K, Bhardwaj AK, Ramamurthy T, Niyogi SK. Triplex PCR assay for the rapid identification of 3 major Vibrio species, Vibrio cholerae, Vibrio parahaemolyticus, and Vibrio fluvialis. Diagn Microbiol Infect Dis 2013;76(4):526-8.
14. Wei S, Zhao H, Xian Y, Hussain MA, Wu X. Multiplex PCR assays for the detection of Vibrio alginolyticus, Vibrio parahaemolyticus, Vibrio vulnificus, and Vibrio cholerae with an internal amplification control. Diagn Microbiol Infect Dis 2014;79(2):115-8.
15. Chuang LY, Cheng YH, Yang CH. Specific primer design for the polymerase chain reaction. Biotechnol Lett 2013;35(10):1541-9.
16. Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 2012;(13): doi:10.1186/1471-2105-13-134.
17. Apte A, Daniel S. PCR primer design. Cold Spring Harb Protoc. 2009;(3): doi:10.1101/pdb.ip65.
18. Dieffenbach CW, Lowe TM, Dveksler GS. General concepts for PCR primer design. PCR Methods Appl 1993;3(3):S30-S37.
19. Kumar A, Chordia N. In silico PCR primer designing and validation. Methods Mol Biol 2015;1275:143-51.
20. Gardes J, Croce O, Christen R. In silico analyses of primers used to detect the pathogenicity genes of Vibrio cholerae. Microbes Environments. 2012;27(3):250–6.
21. Safa A, Nair GB, Kong RY. Evolution of new variants of Vibrio cholerae O1. Trends Microbiol 2010;18(1):46-54.
Recibido: Octubre de 2015 Aceptado: Diciembre de 2015

