










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introducción
El ciclo de vida de un producto transita por el desarrollo farmacéutico, la transferencia tecnológica, la producción y la discontinuidad.1 Es un concepto emitido por la guía Q8 del Consejo Internacional para Armonización de los Requisitos Técnicos de los Productos Farmacéuticos de Uso en Humanos, conocida por las siglas en inglés ICH.2 Esta consideración es válida para la industria biotecnológica cubana que debe acceder a los mercados internacionales con productos de alto valor agregado, que contribuyan a la economía del país y permitan elevar los niveles de salud de la población cubana.
El Centro de Ingeniería Genética y Biotecnología (CIGB) posee un sistema de gestión de la calidad dirigido al control de los proyectos desde la investigación, el desarrollo, la producción y comercialización de ingredientes farmacéuticos activos (IFA) biotecnológicos para uso humano y veterinario, para hacer más viable y efectivo el cumplimiento de las Buenas Prácticas de Fabricación (BPF).
En el CIGB existía un proceso de obtención de una proteína recombinante que se usaba en el sistema diagnóstico de una enfermedad viral crónica, pero el recobrado era bajo y no lograba las especificaciones de calidad requeridas por las regulaciones vigentes para un inmunoterapéutico para uso humano.3,4) Se decidió trabajar en un proyecto de investigación dedicado a lograr un inmunoterapéutico humano, por lo que fue necesario diseñar un nuevo proceso tecnológico para obtener la molécula de interés con las especificaciones de calidad de inmunoterapéutico para administrar a seres humanos.
La transición de la etapa de investigaciones a desarrollo farmacéutico del producto deberá basarse en la administración de riesgo a la calidad tomando en cuenta las experiencias y conocimientos previos, la complejidad del proceso y las especificaciones del producto. La identificación, análisis, evaluación y revisión de los riesgos son necesarios para la toma de decisiones que controlan los gastos, acortan los plazos y logro de objetivos.5 La transferencia de la tecnología de la obtención de la proteína para sistemas diagnóstico de la etapa de investigación hacia la fase de desarrollo tecnológico se realizó cumpliendo con el procedimiento interno y las recomendaciones de las regulaciones.6 En este trabajo se muestra la aplicación de la gestión de riesgos al diseño de un nuevo proceso de producción del IFA de un inmunoterapéutico producido en el CIGB.
Materiales y Métodos
El trabajo en equipo,7 el diagrama de flujo,8) el método de las 6M o análisis de dispersión,9,10 el diagrama causa-efecto10 y la metodología del análisis modal de fallas y efectos (AMFE)10 fueron las técnicas y métodos de calidad empleados en esta investigación.
La administración de riesgos a la calidad para la identificación de los factores que pudieran influir en el diseño del nuevo proceso de producción del IFA de la proteína recombinante como inmunoterapéutico humano fue dirigida por un equipo de nueve integrantes que trabajan en las áreas de Investigaciones, Desarrollo Tecnológico, Gestión de la Calidad y Asuntos Regulatorios (GCAR) y Control de la Calidad del CIGB, según las recomendaciones de varios autores en la literatura consultada.11,12
Los especialistas se seleccionaron por sus responsabilidades en el proyecto de la proteína recombinante para uso diagnóstico, su conocimiento teórico y práctico, tanto en el propio proceso como en BPF, la posibilidad de participación creativa, la capacidad de resolución de problemas, el comportamiento grupal y la orientación y respuesta lógica a las interrogantes que se formularon para definir las acciones que se tomaron.13
Primeramente, se realizó la representación esquemática de la secuencia de las operaciones del proceso de obtención de la proteína con fines diagnóstico utilizando el diagrama de flujo8 (Fig. 1) que sirvió para resumir las características de la tecnología propuesta a transferir de la etapa de investigaciones a la de desarrollo tecnológico.

Para aplicar el trabajo en equipo7 se efectuaron seminarios interactivos en que los investigadores transmitieron a los especialistas de Desarrollo Tecnológico y demás miembros del grupo: la descripción del proceso de obtención de la proteína para uso diagnóstico, la caracterización físico-química de la proteína y los resultados obtenidos en los estudios de inmunogenicidad en animales que demostraban la inducción de la respuesta inmune celular y humoral.
Con el método de las 6M o análisis de dispersión9,10 se identificaron los riesgos del proceso a transferir de la etapa de investigaciones, mientras que el diagrama causa-efecto10 sirvió para la identificación de las posibles causas que pudieran influir en el bajo recobrado del proceso y el incumplimiento con las especificaciones de calidad de un producto biofarmacéutico para administrar en seres humanos (Fig. 2).
La metodología del AMFE10,13,14 se aplicó al proceso de obtención de la proteína recombinante de uso diagnóstico para analizar las fallas potenciales enfocadas en posibles fuentes de riesgos y definidas por tres parámetros: ocurrencia (O), severidad (S) y detección (D). El número de prioridad de riesgo (NPR) se calculó como el producto de los tres factores (NPR=S*O*D).15,16 Los criterios para la clasificación se discutieron con el grupo de expertos y se resumieron en la Tabla 1.
Tabla 1 Criterios para la clasificación de la severidad, ocurrencia y detección.
| Valor | Severidad (S) | Ocurrencia (O) | Detección (D) |
|---|---|---|---|
| 3 | Afecta la calidad del ambiente, producto o consistencia del proceso. | Ha ocurrido más de una vez en el período. | Probabilidad remota o imposible de detectar. |
| 2 | Afectación indirecta sobre la calidad del ambiente, producto o consistencia del proceso. | Ha ocurrido al menos una vez en el período. | Probabilidad baja de detección. |
| 1 | No afecta la calidad del ambiente, producto o consistencia del proceso. No incumple las exigencias regulatorias. | No ha ocurrido antes, pero puede ocurrir. | Probabilidad muy grande de detección. |
Los valores de NPR pueden variar desde 1 (valor mínimo) hasta 27 (valor máximo). Se definieron tres intervalos de NPR para encauzar las decisiones sobre el tipo de acción a tomar, clasificados en: bajo cuando NPR≤3; medio: de 4 a 9 y alto si NPR>9.

Cuando los valores del NPR están en el intervalo: 1 ≤ NPR ≤ 3, no hay impacto del riesgo sobre el proceso y no requiere validación. Si el NPR se encuentra: 4 ≤ NPR ≤ 9 no tiene un impacto significativo sobre la calidad el proceso/producto, pero debe ser documentado. Si NPR > 9 tiene un impacto directo en la calidad del proceso/producto.13
Resultados y Discusión
Identificación, evaluación, control y revisión de los riesgos aplicado al proceso de obtención de la proteína recombinante de uso diagnóstico
En la Tabla 2 se presenta el resultado de la identificación de los principales modos de falla, efectos de las fallas, causas potenciales y NPR acorde al estudio realizado a partir del conocimiento del flujo tecnológico (Fig. 1) y del diagrama causa-efecto (Fig. 2).
La toma de acciones tiene que ser priorizada para las fallas con una ponderación del NPR alto. En primer lugar, se identificaron como riesgos la insuficiente cantidad de viales de los bancos de células y el medio de cultivo complejo que contenía materias primas de origen bovino en su composición. Con este análisis se identificó la incorrecta manipulación de las muestras por los operarios con probable repercusión en el rendimiento esperado del proceso. En los pasos de ruptura y precipitación salina, el uso de un equipamiento no adecuado (no tenía un sistema de enfriamiento incorporado para garantizar la temperatura de la crema), una insuficiente agitación y los parámetros pH y conductividad fuera de límites de especificación pudieron haber provocado afectaciones en el recobrado y en la pureza debido a un alto porcentaje de contaminantes del hospedero como: ADN, proteínas y endotoxinas.
Otras fallas detectadas fueron el empleo de un medio filtrante inadecuado y una incorrecta preparación del sistema de filtración. Las BPF para productos estériles vigentes recomiendan utilizar un filtro estéril de 0,22 micras (o menos) de poro nominal, o con propiedades al menos equivalentes de retención de microorganismos para pasar el producto a un recipiente previamente esterilizado. Además, enuncian que se debe prestar una atención especial para asegurar la esterilidad del producto al ensamblaje del filtro. Todo lo anterior con el propósito de cumplir con uno de los requisitos de calidad de este IFA destinado a la formulación de un fármaco de administración en seres humanos.17
La gestión de riesgo se realizó por anticipado para diseñar el nuevo proceso de producción del biofarmacéutico para uso terapéutico en pacientes; se identificaron las posibles causas de las fallas potenciales con mayor influencia en la tecnología existente:
1. Cantidad insuficiente de viales de los Bancos de Células Primario (BCP) y de Trabajo (BCT) para un proceso consistente. Los bancos de células confeccionados en el área de Investigaciones no se habían realizado por duplicado, las condiciones de almacenamiento no eran adecuadas para garantizar la viabilidad de las células y, al mismo tiempo, protegerlas de la contaminación. Por otra parte, el control periódico de los bancos no se había realizado para determinar su disponibilidad para el uso.3,4
Tabla 2 Resultados del Análisis Modal de Fallos y Efectos del nuevo proceso del inmunoterapéutico.
| 6M | Escenario de riesgo | O | S | D | NPR | Acciones | O | S | D | NPR |
| Mano de obra | Incorrecta preparación de los bancos de células | 3 | 2 | 1 | 6 | Capacitación del personal y supervisión del cumplimiento de los procedimientos | 3 | 1 | 1 | 3 |
| Inadecuada preparación de medios de cultivo y soluciones | 3 | 3 | 1 | 9 | 3 | 2 | 1 | 6 | ||
| Incorrecta inducción del cultivo | 3 | 2 | 1 | 6 | 3 | 1 | 1 | 3 | ||
| Incorrecta manipulación de las muestras | 3 | 2 | 3 | 18 | 3 | 2 | 1 | 6 | ||
| Incorrecta manipulación de las operaciones | 3 | 2 | 1 | 6 | 3 | 2 | 1 | 6 | ||
| Incorrecta preparación del sistema de filtración | 3 | 2 | 3 | 18 | 3 | 2 | 1 | 6 | ||
| Insuficiente capacitación del personal | 3 | 3 | 1 | 9 | 3 | 1 | 1 | 3 | ||
| Maquinaria | Insuficiente agitación | 3 | 2 | 2 | 12 | Comprobar el parámetro: agitación. | 3 | 3 | 1 | 9 |
| Equipamiento no adecuado (sin sistema de enfriamiento) | 3 | 3 | 2 | 18 | Disponer de un sistema de enfriamiento apropiado para garantizar la temperatura de la crema. | 3 | 1 | 1 | 3 | |
| Medio filtrante inadecuado | 3 | 3 | 3 | 27 | Disponer del medio filtrante idóneo | 3 | 1 | 1 | 3 | |
| Metodología | Irregularidades en la higienización del área de trabajo | 3 | 2 | 1 | 6 | Capacitación del personal y supervisión del cumplimiento de los procedimientos | 3 | 1 | 1 | 3 |
| Inadecuada esterilización de los materiales | 3 | 2 | 1 | 6 | 3 | 1 | 1 | 3 | ||
| Inadecuado manejo de las BPF durante la aprobación de los bancos de células | 3 | 3 | 1 | 9 | 3 | 1 | 1 | 3 | ||
| Pasos no cromatográficos y cromatográficos incapaces de remover los contaminantes del hospedero | 3 | 3 | 3 | 27 | Diseño e implementación de un nuevo proceso para disminuir el alto porcentaje de contaminantes | 3 | 1 | 1 | 3 | |
| Materiales | Insuficiente cantidad de viales de los bancos de células | 3 | 3 | 3 | 27 | Elaborar los Banco de Células Primarias y Banco de Células de Trabajo con un número suficiente de viales. | 3 | 3 | 1 | 9 |
| Disminución de la estabilidad plasmídica | 3 | 1 | 2 | 6 | Comprobación periódica de los Banco de Células Primarias y Banco de Células de Trabajo. | 3 | 1 | 1 | 3 | |
| Vector de expresión deteriorado | 3 | 1 | 2 | 6 | 3 | 1 | 1 | 3 | ||
| Materias primas, matrices o soluciones vencidas | 3 | 1 | 3 | 9 | Verificar la fecha de vencimiento de las materias primas y materiales. | 3 | 2 | 1 | 6 | |
| Medio de cultivo complejo con materias primas de origen bovino | 3 | 3 | 3 | 27 | Utilizar medios de cultivo químicamente definidos. Verificar si los proveedores de materias primas emiten certificados libres de encefalopatía espongiforme bovina/encefalopatías transmisibles. | 3 | 2 | 1 | 6 | |
| Alto porcentaje de contaminantes del hospedero | 3 | 2 | 3 | 18 | Capacitación y supervisión del personal en el manejo de las muestras | 3 | 2 | 1 | 6 | |
| Medio ambiente | Temperatura de trabajo del área no adecuada | 3 | 3 | 1 | 9 | Supervisión permanente automatizada de la temperatura del local Supervisión de la limpieza de área y cabinas de flujo laminar | 3 | 1 | 1 | 3 |
| Inadecuada limpieza de área y cabinas de flujo laminar | 3 | 2 | 1 | 6 | 3 | 1 | 1 | 3 | ||
| No existen condiciones adecuadas de cultivo durante el inóculo | 3 | 2 | 1 | 6 | Revisar periódicamente el programa de mantenimiento preventivo del equipamiento Revisar periódicamente el programa de monitoreo ambiental del área | 3 | 1 | 1 | 3 | |
| Microbiología del área fuera de especificación | 3 | 3 | 1 | 9 | 3 | 1 | 1 | 3 | ||
| Medición | Técnicas analíticas no validadas | 3 | 2 | 1 | 6 | Validar las técnicas analíticas | 3 | 1 | 1 | 3 |
| Parámetros de operación fuera de límites de especificación (pH y conductividad) | 3 | 3 | 2 | 12 | Establecer un punto de inspección para pH y conductividad | 3 | 2 | 1 | 6 | |
| Uso de instrumentos de medición no calibrados | 3 | 3 | 2 | 12 | Revisar periódicamente el estado de la calibración de los instrumentos | 3 | 1 | 1 | 3 |
S: severidad. O: ocurrencia. D: detección. BCP: Banco de células primario. BCT: Banco de células de trabajo. MP: materia prima. EEB: encefalopatía espongiforme bovina. ET: encefalopatías transmisibles. CLAR: Cromatografía líquida de alta resolución
2. El medio de cultivo utilizado en la preparación de los bancos de células, multiplicación y fermentación de la cepa hospedera fue Luria Bertani (LB).18,19 Este tipo de medio complejo es caro, los microorganismos presentan un crecimiento bajo en él y contiene materiales biológicos de origen bovino de proveedores que sus certificados no especifican si los productos están libres de encefalitis espongiforme bovina (EEB) o encefalopatías transmisibles (ET).20
3. La etapa de cosecha, lavado de la biomasa y ruptura tenía bajo recobrado. El equipamiento no era adecuado (sin sistema de enfriamiento incorporado) para obtener el rendimiento de proteína esperada. Además, las condiciones establecidas a nivel de laboratorio (ruptura celular con ultrasonido) no podían ser reproducidas para su futura transferencia tecnológica al sistema productivo. Estas operaciones con estos equipos no aseguraban que el producto intermedio se recuperase con consistente calidad.3
4. La etapa de purificación no rendía los niveles de pureza y recobrado esperados para el IFA de un inmunoterapéutico humano. Los recobrados eran bajos en los pasos cromatográficos y no cromatográficos, pues el proceso no removía suficientemente los contaminantes del hospedero, tales como ADN, proteínas y endotoxinas, lo que no cumplía con los requisitos de las BPF de productos biológicos vigentes para la producción de proteínas recombinantes que recomiendan procesos de purificación capaces de eliminar las proteínas no deseadas de la célula huésped, los ácidos nucleicos, contaminantes del hospedero, carbohidratos, virus y otras impurezas que estarán dentro de los límites definidos validados.4
5. El purificado final no cumplía con las especificaciones de calidad de un producto biofarmacéutico estéril. La filtración del purificado final utilizaba un medio filtrante inadecuado (membrana plana de 0,2 µm) y la operación no cumplía con las condiciones sanitarias ni con los principios de BPF exigidos para un IFA destinado a un farmacéutico estéril.17
Las causas potenciales aportaron una parte fundamental a la variabilidad y la calidad del proceso, por lo que los esfuerzos de mejora se enfocaron hacia estos elementos en conjunto con las acciones enfocadas a la mitigación de los riesgos.
La estrategia a seguir para establecer el nuevo proceso de producción de IFA del inmunoterapéutico presentó las siguientes acciones:
1. Elaborar los BCP y BCT con un número suficiente de viales, según el procedimiento interno, que prevé los ensayos periódicos de caracterización: viabilidad, pureza, análisis de restricción, estabilidad plasmídica y expresión de la proteína recombinante como parte de los requisitos exigidos para declararlos como aptos para su uso.3,4
2. Utilizar medios de cultivo de composición definida en la fermentación, los cuales se preparan a partir de sales inorgánicas y una única fuente de carbono.
3. Establecer en el proceso de cosecha, lavado y ruptura de la biomasa el equipamiento adecuado para incrementar el recobrado de la etapa.
4. Implementar mejoras en los pasos cromatográficos y no cromatográficos para aumentar la pureza de la proteína y disminuir los contaminantes procedentes del hospedero: ADN, proteínas y endotoxinas.
5. Emplear en la filtración del purificado final un medio filtrante adecuado y hacer el chequeo de control microbiológico antes y después al material filtrado para conocer si cumple con las especificaciones de un producto biofarmacéutico estéril.
También se formularon otras acciones para controlar riesgos que afectaban la obtención de la proteína con la pureza requerida para un producto inmunoterapéutico estéril como: establecer el chequeo de las variables, pH y conductividad en los pasos de ruptura celular, precipitación salina, intercambio iónico y cromatografía líquida de alta resolución -gel filtración (CLAR-GF). Además, implantar el control de los parámetros críticos (pH y presión) durante la fermentación; disponer de un sistema de enfriamiento apropiado para garantizar la temperatura de la crema rota; así como, entrenar y supervisar el personal de operaciones de apoyo y proceso. Dichas condiciones y controles de proceso durante el crecimiento celular, la expresión y purificación de proteínas se deben mantener dentro de los parámetros validados, para asegurar un producto consistente con un rango definido de impurezas que esté dentro de la capacidad del proceso para reducirlas a niveles aceptables.4
El propósito de establecer el nuevo proceso del IFA llevó a tomar medidas alineadas con las recomendaciones de las directrices sobre BPF de productos farmacéuticos vigentes en Cuba como:
- Comprobar la certificación de liberación y periodo de vigencia de las materias primas, matrices cromatográficas y soluciones por una persona u organización autorizada, pues todos los materiales que ingresen a la entidad serán sometidos a cuarentena inmediatamente después de su recepción, hasta que sean liberados para su uso; luego deben ser almacenados en condiciones apropiadas establecidas por el fabricante y en un orden tal que pueda efectuarse la segregación de los lotes y la rotación de las existencias, según la regla de que los primeros que expiran son los primeros que salen.21) En los procesos de purificación, las matrices cromatográficas dedicadas a un solo producto, se esterilizarán o higienizarán para los distintos lotes. Se evitará el uso de una misma matriz en diferentes etapas del proceso de producción. Se definirán los criterios de aceptación, las condiciones de operación, la vida útil y los métodos de regeneración, higienización y esterilización de las mismas. También se prestará particular atención a la vigilancia de las cargas biológicas y endotoxinas.4
- Validar las técnicas analíticas utilizadas en las actividades de control pues todos los procedimientos analíticos empleados para análisis deben ser adecuados para el uso al que están destinados. Esto se demuestra por validación de acuerdo al protocolo, que incluye las características de desempeño analítico a ser verificadas para los diferentes tipos de procederes analíticos. Las características típicas que deben considerarse: exactitud, precisión, robustez, linealidad e intervalo, especificidad, paralelismo, límite de detección y de cuantificación.22
- Elaborar un procedimiento para la preparación y esterilización de materiales que tengan efecto para prevenir la contaminación que pueda alterar la calidad de los productos intermedios o del IFA en la esterilidad del producto.17
- Revisar periódicamente el programa de mantenimiento preventivo del equipamiento. Debido a que los servicios de mantenimiento y calibración de los equipos e instrumentos de medición, registro y control se deben realizar a intervalos predefinidos, manteniéndose registros de dichas operaciones. El personal para asegurar el adecuado funcionamiento de los equipos debe revisarlos diariamente o antes de su uso. El programa permanente desde su implementación inicial será revisado como mínimo anualmente.22
- Supervisión automatizada permanente al monitoreo de la temperatura, humedad relativa y presión absoluta de cada local. Los sistemas de calentamiento, ventilación y acondicionamiento del aire, estarán diseñados, construidos y mantenidos para garantizar una temperatura y humedad relativa satisfactoria, minimizar el riesgo de contaminación cruzada entre las diferentes áreas de producción y tener en cuenta la comodidad del personal que trabaja con vestimenta protectora. Es posible que las unidades manejadoras de aire sean específicas para un área. En caso de la emisión de alertas en función de las especificaciones de área establecidas y aprobadas para cada local las causas de los incumplimientos de estos parámetros se investigarán para dictar las acciones aplicables.4
A partir de las fallas detectadas por la gestión de riesgos a la calidad se implementaron mejoras con respecto a la tecnología procedente del área de Investigaciones que se destacan en la Tabla 3. Con estos cambios se diseñó el nuevo proceso productivo del IFA del inmunoterapéutico (Fig. 3) con el propósito de garantizar la calidad del producto y la seguridad del paciente.
Tabla 3 Mejoras en el nuevo proceso de producción del IFA del inmunoterapéutico.
| Proceso de obtención proteína-diagnóstico (Investigaciones) | Nuevo proceso de producción IFA-inmunoterapéutico (Desarrollo Tecnológico) | |
|---|---|---|
| Etapas del proceso | BCP | BCP, BCPE y BCT (suficiente cantidad de viales) |
| Inoculación del BCP en medio LB con kanamicina + triptófano | Inoculación del BCP en medio químicamente definido (materias primas libres de EEB o ET) | |
| Ruptura celular con ultrasonido | Ruptura celular con un homogenizador de alta presión | |
| Precipitación salina diferencial (sulfato de amonio al 10% y 40%) | Precipitación salina diferencial (sulfato de amonio al 20%) | |
| Resuspensión del precipitado-Concentración por ultrafiltración | - | |
| Cromatografía por gel filtración | Cromatografía de interacciones hidrofóbicas (remoción del ADN contaminante del hospedero) | |
| Cromatografía de intercambio aniónico con lavado de tritón X-114 (reducción del contenido de endotoxinas) | ||
| Concentración/diafiltración en sistema de filtración tangencial | ||
| CLAR-GF (pureza ≥ 95%) | ||
| Filtración final esterilizante por membrana plana de 0,2 µm | Filtración final esterilizante por cápsulas | |
| Recobrado | 47% | ≥ 70% |
| Pureza | 90% | ≥ 95% |
BCP: Banco de células primario. BCPE: Banco de células primario extendido; BCT: Banco de células de trabajo. EEB: encefalopatía espongiforme bovina. ET: encefalopatías transmisibles. CLAR-GF: Cromatografía líquida de alta resolución-gel filtración.
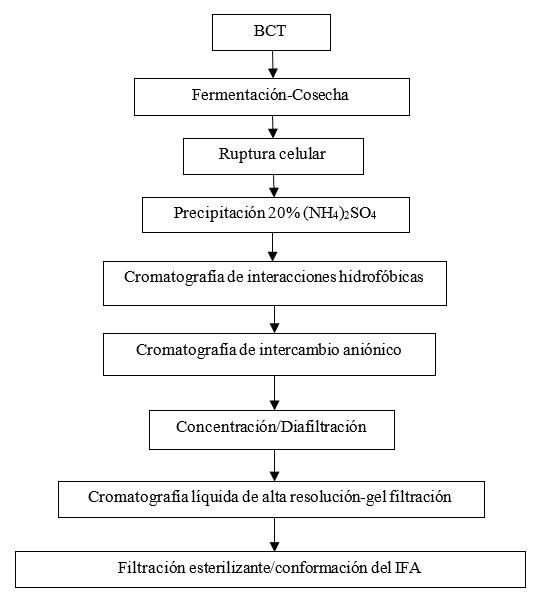
El nuevo proceso de producción rindió el IFA del inmunoterapéutico con las características y atributos críticos de calidad para uso humano que se muestran en la Tabla 4.
Tabla 4 Especificaciones de calidad del IFA inmunoterapéutico para uso humano.
| Característica | Atributo critico de calidad | |
|---|---|---|
| Identidad antigénica | Identificado | |
| Pureza | SDS-PAGE | ≥ 95% |
| CLAR-GF | ≥ 95% | |
| Esterilidad | Pasa la prueba | |
| Endotoxinas | ≤ 1,15 UE/µg | |
| Concentración de proteínas | ≥ 0,30 mg/mL | |
| Proteínas contaminantes de hospedero | ≤ 0,80% | |
| ADN contaminante | ≤ 2 µg ADN/100 µg dosis | |
| pH | 6,3 - 7,3 | |
SDS-PAGE: electroforesis en gel de poliacrilamida en presencia de SDS. CLAR-GF: Cromatografía líquida de alta resolución-gel filtración.
Los atributos críticos de calidad establecidos para la producción de los IFA para uso humano se correspondieron con los requisitos exigidos por las regulaciones nacionales e internacionales.2,3,4
El seguimiento anual al plan de manejo de los riesgos conducido por la Dirección de GCAR, acorde a las recomendaciones de la guía Q9,15 comprobó el estado de cumplimiento de las acciones con una disminución de un nivel de riesgo alto a moderado y bajo (Tabla 2). De la revisión, las causas potenciales de esos riesgos en el nuevo proceso de producción del IFA del inmunoterapéutico humano son atendidas en la fabricación de lotes.
Conclusiones
La gestión de riesgos llevó a mejoras para reducir las fallas detectadas en la tecnología propuesta a transferir que permitió el diseño del nuevo proceso de producción del IFA con el propósito de obtener un inmunoterapéutico para enfermedades virales crónicas. La revisión sistemática y periódica de este análisis de riesgo debe servir como estrategia en el diseño de nuevas producciones biotecnológicas en el CIGB.

