










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBiotecnología Aplicada
versión On-line ISSN 1027-2852
Biotecnol Apl v.26 n.1 La Habana ene.-mar. 2009
REVIEW
Complexity and solutions to the isolation problem of Gram negative lipopolysaccharides' bacteria molecular species
Complejidad y soluciones al problema de aislamiento de las especies moleculares de los lipopolisacáridos de las bacterias Gram negativas.
Elder Pupo1, Eugenio Hardy2
1Laboratorio de Polímeros, Instituto de Ciencia y Tecnología de los Materiales, Universidad de La Habana. Cuba
2División de Química-Física, Centro de Ingeniería Genética y Biotecnología, CIGB. Ave. 31 entre 158 y 190, Cubanacán, Playa, AP 6162, CP 10600, Ciudad de La Habana, Cuba
ABSTRACT
Gram negative bacteria frequently synthesize a great number of lipopolysaccharides (LPS) molecular species. The high LPS structural heterogeneity and its trend to conform molecular aggregates make the fraction of its molecular species extremely difficult. The application of chromatographic methods individually has allowed the obtainment of little homogeneous LPS fractions. With the combination of orthogonal chromatographic principles, only the purification of the simplest rough type LPS up to chemical homogeneity has been reached. This has determined that efforts aimed at more homogeneous LPS isolation is focused on the use of electrophoresis in slab gel. Electrophoresis methods are capable of separating species of similar molecular masses next to from the most complex smooth type LPS. Consequently upon this basis, a new methodology allowing the isolation of intact LPS of either rough or smooth types up to electrophoretic homogeneity has been developed. Under such methodology, sensible structural analysis through mass spectrometry and LAL (Limulus Amebocyte Lysate) activity of LPS individual species is feasible, composed of just a few chemical species. This methodology has proved the possible way of combining electrophoresis with other orthogonal separation principles, a further step which could guarantee the obtainment of chemically homogeneous preparations from complex LPS mixtures.
Keywords: lipopolysaccharides, isolation, chromatography, electrophoresis, reverse staining, elution, mass spectrometr.
RESUMEN
Las bacterias Gram negativas sintetizan de manera frecuente un gran número de especies moleculares de lipopolisacáridos (LPS). La elevada heterogeneidad estructural de los LPS y su tendencia a formar agregados moleculares hacen extremadamente difícil el fraccionamiento de sus especies moleculares. La aplicación de métodos cromatográficos de forma individual ha permitido la obtención de fracciones de LPS poco homogéneas. Con la combinación de principios cromatográficos ortogonales, solo se ha logrado la purificación hasta homogeneidad química de los LPS de tipo rugoso más simples. Ello ha determinado que los esfuerzos dirigidos al aislamiento de LPS más homogéneos se centren en el uso de la electroforesis en el gel plano. Los métodos de electroforesis son capaces de separar especies de masas moleculares cercanas de LPS más complejos de tipo liso. Consecuentemente, sobre esta base se ha desarrollado una nueva metodología que permite el aislamiento de LPS intactos de tipo rugoso o liso hasta homogeneidad electroforética. Con ella es posible el análisis estructural sensible por espectrometría de masas y la medición de la actividad LAL (Limulus Amoebocyte Lysate) de especies individuales de LPS, compuestas de unas pocas especies químicas. Esta metodología ha abierto las puertas hacia la posibilidad de combinar la electroforesis con otros principios ortogonales de separación, un próximo paso que podría facilitar la obtención de preparaciones químicamente homogéneas a partir de mezclas de LPS complejas.
Palabras clave: lipopolisacárido, aislamiento, cromatografía, electroforesis, tinción inversa, elución, espectrometría de masas.
INTRODUCTION
Lipopolysaccharides (LPS), also called endotoxins, are major components of the Gram negative bacteria outer membrane. Generally, LPS are formed by a lipidic domain named lipid A which functions like an anchor at the outer membrane and is bound to an oligopolysaccharide extending towards the exterior part of the bacteria. A very same bacterial strain frequently synthesizes a great number of LPS molecular species.
LPS as integral components of the outer membrane fulfill essential functions for the bacteria and show a wide variety of both, immunological and biological activities in other organisms (1, 2). These glycolipids play a key role of mediators in the bacterial infection, sepsis, and septic shock (1, 2). LPS are considered targets, as these biomolecules and their synthetic derivates are used as antigens or adjuvants to provide vaccines against diverse pathogens (3-5).
In this review, several aspects of LPS molecular species separation are examined. First, the complexity of LPS mixtures synthesized by the bacteria is under discussion: the great amount of subspecies, the high number of chemical structures, and the differences in biological activity. Next, the need for the separation of subspecies for the improvement of LPS biochemical characterization is fundamented. Finally, an analysis on advances in the isolation of molecular species and problems arising from the methods used for such purpose is performed, from the point of view of the homogeneity of the preparations, its applicability to different LPS sources, and compatibility with standard methods for LPS biochemical analysis.
STRUCTURAL HETEROGENEITY OF LIPOPOLYSACCHARIDES
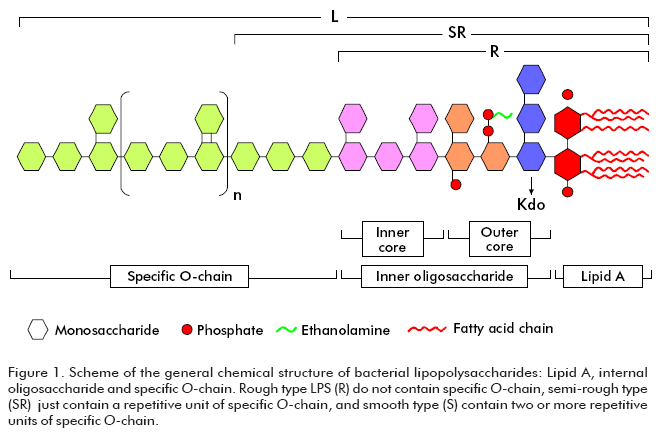
In general, LPS chemical structure (Figure 1) comprises three regions: a lipid (called lipid A), an internal oligosaccharide bound to lipid A by Kdo (3-deoxi-Dmanno- octulosonic acid) and specific O-chain bound to the internal oligosaccharide. Specific O-chain, consisting of a repeated sequence of just a few monosaccharides, presents a variable polymerization degree. Consequently, some LPS do not contain specific O-chain (known as rough type LPS), which only show a single repetitive unit (semi-rough type; Figure 1), and others showing several repetitive subunits in the specific O-chain (smooth type). In spite of LPS having the biological activities associated to the molecule lipidic region, the polysaccharide also plays a significant functional role (7-10).
It is fairly common that LPS produced by a bacterial strain differ in respect to: i) the number, nature, length, and the position of fatty acids from lipid A; ii) the replacement of phosphate groups and phosphorilation degree of lipid A; iii) length sequence, and oligopolysaccharide sugars; or iv) the replacement of the oligopolysaccharide with acetyl groups, phosphate and phosphorilethanolamine. As a result, the number of LPS molecular species synthesized by a bacterial strain is usually big. For example, the typical laddering pattern shown by smooth type LPS, through the electrophoresis technique of polyacrylamide gel in presence of dodecyl sodium phosphate (SDS) or sodium deoxycholate (DOC), reveals multiple LPS populations with a different polymeration degree of specific O-chain (

LPS variants may also differ notably in their biological activity in in vivo and in vitro assays. Natural occurrence of variations in the chemical structure of lipid A, responsible for most LPS biological activity (12) is supposed to induce remarkable differences among the biological activities of LPS molecular species. This may be inferred from the structure-function relations of lipid A which have been established in biological studies with lipid A obtained from natural sources or their synthetic structures (13-16).
Apart from the lipid A region, structural heterogeneity associated to the polysaccharidic-O-chain constitutes other variation source of biological activity for LPS species. For instance, the activation mechanism of macrophages or similar cells by smooth type LPS may be different from that of rough type LPS (17). Both LPS show distinctive qualitative differences according to their interactions with the complement system and a different distribution in organs in vivo. While hepatic adsorption of smooth LPS from blood occurs in an exclusive manner by sinusoidal cells (Kupffer cells); rough type LPS absorption occurs through the Kupffer cells themselves and hepatocytes (18). Smooth and rough type LPS interactions with the high density lipoprotein in vivo are also different. As a result, great amounts of smooth type LPS accumulate at the suprarenal glands, while the amounts of rough type LPS in this organ are insignificant (19).
The different LPS molecular forms may also differ in respect to the cell activation mechanism. In recent studies with wild type LPS strains and Salmonella and Escherichia coli mutants of rough phenotype, it was determined that rough type LPS easily activate the cells expressing the LPS signaling receptor, composed by the Toll-like receptor 4, and the myeloid differentiation protein 2 (TLR4/MD-2). Smooth type LPS require LPS binding proteins (LBP and CD14) in order to activate this same receptor (20). This conclusion is derived from an in vitro LPS activation experiment of mastocytes not expressing the membrane protein CD14. Rough type LPS was a potent inducer of interleukin-6 and the tumor necrosis factor-α; however, smooth type LPS lacked from enhancing activity (20). As a result of this finding, it was determined in physiological conditions that rough type LPS activates a higher cell spectrum than smooth type LPS. In addition, as wild bacteria LPS (smooth phenotype) also contain a fraction of rough type low molecular mass LPS; it is speculated that the contribution of rough type LPS to the immune response and the physiopathology of the infection provoked by Gram negative bacteria could be higher than the smooth type LPS forms (20).
RELEVANCE OF SEPARATION METHODS FOR THE BIOCHEMICAL ANALYSIS OF LIPOPOLYSACCHARIDES
Facing a so complex LPS molecule nature demands the use of sophisticated analytical techniques and frequently of different biochemical methods for its structural characterization. Nuclear magnetic resonance and mass spectrometry (MS) are complemented to determine the most representative LPS structures. However, analyses on LPS are usually being performed obtained in high scale-grown bacteria, as significant amounts are required. This hinders LPS characterization when such amounts are too scarce (e.g., in direct samples taken from infected patients). LPS analyses of clinical samples, from bacterium grown in a culture medium undertakes the inconvenient that such biomolecules may vary its structure during cell culture, a kind of phenomenon called phase variation occurring due to the use of alternative oligopolysaccharides biosynthesis pathways (21).
The sensibility of LPS structural characterization methods has considerably increased right from the use of a separation technique, the O-desacylated LPS capillary electrophoresis, in combination with MS (22). This methodology has allowed the analysis of so small amounts of LPS as the ones expressed by a few bacterial colonies (23).
Nevertheless, the LPS structural characterization highest handicap is in its structure which is often inferred from the analysis of its fragments (O-desacylated LPS, lipid A, oligopolysaccharide), in different hydrolysis reactions. Due to certain hydrolysis conditions may also induce to the loss of replacement groups bound to the glycosidic skeleton, the full natural heterogeneity of these molecules cannot be established.
A solution to this problem lies in the direct structural analysis of intact LPS species. Plasma desorption MS, and further matrix-assisted laser desorption/ ionization time-of-flight MS (MALDITOFMS) has allowed the structural analysis of intact, low molecular mass rough type LPS molecules (24). However, the amphipathic nature and the high heterogeneity of smooth type LPS show a series of wide ion peaks too little resolved (25, 26). It is only possible to know mass difference among the main components and elucidate the mass of specific O-chain repetitive unit starting from those spectra. The complex spectrum of smooth type LPS does not allow to obtain any kind of information on lipid A composition (12). Consequently, the determination of lipid A structural variability has not been possible; the most important LPS component from the biological point of view among the smooth type individual glycoforms. In very few studies, average differences among lipid A acylation patterns, from either long or short polysaccharidic chains of LPS species though with little resolution, have been known only after the fractioning of LPS mixtures (27, 28). For instance, the analysis by plasma desorption MS of two chromatographic fractions, yet heterogeneous, of low molecular mass (rough type) and high molecular mass (smooth type) LPS of E. coli 097, showed that rough type LPS fraction is found tetra-acylated while the smooth type is found hexaacylated (28). In a different study, smooth type LPS from Salmonella abortus equi has a lesser acylation degree than the rough type fraction (27). All the above proves that the use of high resolution separation methods preserving LPS chemical integrity and its combination with the most sensitive structural analysis techniques (e.g., MALDITOF-MS, ESI-MS) may contribute to a wider knowledge on the native chemical structure of these biomolecules.
Huge difficulty to separate LPS has provoked that so far, most biological studies of LPS isolated from natural sources have defined the biological activities of these glycolipids from heterogeneous preparations. In very few cases, fully homogeneous preparations, such as LPS Re type from Rhodobacter sphaeroides (29), have been used. As a result, in most studies, the direct biological activity of different LPS molecular species have not been possibly assessed, nor derived the precise relations between the structure and function of these biomolecules. Hence, the development of methods allowing the isolation of LPS individual glycoforms up to homogeneity is a ruling objective, as it may notably contribute to the determination of the biological properties of these macromolecules.
SOLUTIONS FOR THE ISOLATION OF LIPOPOLYSACCHARIDE MOLECULAR SPECIES
LPS fractioning in their molecular species at semipreparative or preparative scale is extremely difficult due to their high heterogeneity and trend to form molecular aggregates. The presence of lipidic structures, both polar and ionic in LPS make these glycoconjugates be very amphipathic and little soluble in water or most organic solvents (6). For instance, smooth type LPS critical aggregation concentration of E. coli 0111:B4, E, coli 055:B5, S. abortus equi, Salmonella typhimurium and Serratia marcescens were found between 10 and 38 mg LPS/mL (30). LPS aggregation also produces big and polydisperse multimolecular entities of molecular mass between 105 and 106 Da (31, 32). Consequently, LPS separation under diverse principles has been combined with methods allowing its disaggregation.
Afterwards, diverse LPS isolation methods are analyzed and described; as a lot of emphasis is made on the performance of the following parameters: i) LPS purity; ii) preservation of the molecule chemical integrity; iii) applicability to diverse LPS sources; and iv) compatibility with biological and structural LPS characterization methods.
Liquid-liquid extraction
At least 18 different methods for bacterial LPS extraction have at least been described. Among them: the hot phenol-water (33), phenol-chloroform-petroleum ether (34), and the Hitchcock-Brown methods stand as the mostly used, including the extraction with SDS and digestion with proteinase K (35). The result of the application of these methods lies on LPS separation of other biomolecule types (e.g., proteins, DNA) conforming the microorganism. Generally, obtained preparations are highly heterogeneous in terms of LPS molecular species, but relatively pure regarding the content of proteic contaminants, DNA and phospholipids. As an initial purification stage, liquid-liquid extraction is useful, as it facilitates the further separation of LPS molecular species through the use of other techniques.
Liquid chromatography
Several chromatographic methods for the separation of LPS species have been used: countercurrent chromatography (36), gel filtration, ionic exchange (38), hydrophobic interactions (39) using silica gel (40), hydroxyapatite (41), and reverse phase (42, 43). Despite the existing variety of separation principles in which they are based, they generally produce nonhomogeneous LPS fractions.
For example, countercurrent chromatography, based on the differential partition of smooth and rough LPS between the organic and aqueous phase, is the one which has been used to fractionate LPS from wildtype strains of S. enterica, S. serovars abortus equi and Salmonella friedenau and Citrobacter freundii. The two fractions contain various LPS species of long polysaccharidic chain or not containing specific Ochain (36). On the other side, gel filtration chromatography Sephadex G-200, or Sepharose 4B in presence of SDS or DOC, which has been much used for LPS separation according to molecular mass, generally produces between two and three lipopolysaccharide heterogeneous fractions (37).
Hydrophobic interaction chromatography in octylsepharose gels, of adsorption in silica gel or in hydroxyapatite gels, were used in slab type LPS separation of S. typhimurium and S. abortus equi; Yersinia enterocolitica O:11,17, E. coli O97 and S. typhimurium LT2; or from various Bordetella pertussis strains, respectively. LPS separated into two types of fractions containing smooth and rough types of LPS populations (28, 39, 40).
Ionic exchange chromatography in tertiary amine anionic exchangers (e.g., Amberlite XE 220 resin) has been used to separate native LPS (38). Although obtained fractions had different demonstrated biological and structural properties, purity could not be demonstrated (e.g. through a higher LPS separation power method, as SDS-PAGE). This chromatographic method has also the inconvenient of using a strong alkaline elution agent, capable of modifying LPS properties and not allowing the recover of some of the LPS fractions from the chromatographic column due to the very high affinity by the resin.
High-resolution reverse phase liquid chromatography (RP-HPLC) has been used to purify LPS from Klebsiella pneumonae O1 K2 homogenates. Through this method, a protein-free but heterogeneous LPS preparation was obtained, with the same biological properties than the one extracted with the hot phenolwater method (41).
Thin layer chromatography (TLC) is one of the simplest and fastest methods used for the separation of glycolipids. This has been recently used for the direct micro extraction and analysis of rough type native LPS through MALDI-MS (44). However, TLC has a very limited resolution for native LPS, and has proved only been useful in the partial separation of LPS species of low molecular mass and little complexity. Difficulties in LPS extraction of TLC plates due to the contamination of these with silica gel and a low LPS recovery have also been found (45).
Summarizing, completely homogeneous preparations have not been reached with any of the chromatographic methods been used individually for the purification of LPS molecular species. This conclusion has suggested that, perhaps, the combination of various orthogonal principles could be needed in order to be able to separate the different LPS heterogeneities (e.g.,electric charge, hydrophobicity, molecular size).
Such idea has been demonstrated with the purification up to homogeneity of the simplest LPS (Re type, consisting in lipid A bound to two units of Kdo) of E. coli D31m4, by combining ion-exchange chromatography and RP-HPLC (46). LPS extracted from the rough mutant Re from E. coli D31m4, was disaggregated with 0.1 M EDTA and fractioned in a diethylaminoethyl-cellulose. The biphosphorylated form of the lipopolysaccharide was obtained at this chromatographic step, separated from the phospholipids and other charged LPS forms. In order to separate the majoritary hexacylated form from others present in this preparation, as the pentacylated one, a RPHPLC (C18) was performed. Native glycolipids were separated in this new chromatography with the use of tetrabutylammonium salt, an LPS disaggregating agent. When it was analyzed by MS, purified LPS proved fully homogeneous. In spite, this result evidenced for the first time that it is possible to isolate LPS to homogeneity, the purification procedure was only applied to the simplest LPS, Re type, and has not successfully been used in the separation of other more complex LPS types (e.g., smooth type LPS).
Electrophoresis
Capillary electrophoresis
For the structural LPS characterization, one of the modern separation methods widely used is capillary electrophoresis in combination with MS (22, 47). Separation through capillary electrophoresis resolves very efficiently O-desacylated rough type LPS species.
For instance, it allows the separation of glycoforms that differ only in just a phosphate group (48). When connected in line with MS, ESI-MS is a structural analysis method of a very high resolution and sensitivity (48, 49). However, this technique is limited by the release of some proportions of phosphate groups and phosphoethanolamine during LPS O-desacylation with alkaline media (50, 51). Thus, a non-natural LPS heterogeneity is introduced, which structure is more difficult to be analyzed.
Intact LPS, though, has not successfully been separated or completely analyzed equally through capillary electrophoresis because LPS is too little soluble in the buffer solutions used in this technique. On the other hand, the separation of native smooth type LPS in presence of detergent (e.g., SDS) shows low resolution (52). Capillary electrophoresis has not been used for the isolation of native LPS; either its characterization in biological activity assays.
Electrophoresis in polyacrylamide gel of cylindrical format in presence of detergents
Preparative DOC-PAGE electrophoresis of cylindrical format has successfully been used for the separation of different LPS glycoforms of various microorganisms with a high purity (53, 54). Apart from the different LPS types (rough, semi-rough and smooth) diverse LPS species of different molecular sizes of smooth or semi-smooth types are separated (53). However, the usefulness of this format for the biological and structural characterization of individual LPS species has not been fully demonstrated yet. In fact, purified individual species were mixed again in heterogeneous fractions (rough or smooth types) in order to determine their monosaccharide composition (54). This technique disadvantageously lacks of a method for in line detection of electroeluted LPS. Consequently, LPS fractioning is difficult and imprecise, and may eventually provoke the mixing of LPS species which have priorly been separated in the gel. On the other side, LPS elutes in presence of detergents and salts interfering with the further analysis of these molecules by biological assays or MS.
Electrophoresis in slab-polyacrylamide gel in presence of detergents
Systematic studies on LPS molecule separation by slabpolyacrylamide gel electrophoresis (slab-PAGE), performed through the use of LPS chemotypes characterized in chemical terms (e.g., Salmonella (35)), showed how differences in LPS separation profiles detected with silver (55) correlated with the differences in chemical structures among LPS. In fact, smooth type glycolipids, only differing in O-chain sugar components separated themselves in a laddering differential pattern (35). Likewise, LPS band mobility of the highest migration of bacterial mutant chemotypes (Rd, Rc, Rb and Ra) increases while the inner oligosaccharide shortens (35). On the other hand, inconsistencies in separation (like the diffuse, low migration speed, doublet shaped bands produced as a result of the incomplete dissociation of LPS aggregates during electrophoresis) have resolved by electrophoresis of the gel before separation or by increasing SDS concentration in it (56). In order to solve this problem, SDS was replaced with sodium desoxycolate, which dissociated with higher efficiency LPS multimers and generated well solved smooth and rough type LPS patterns (57). A higher LPS resolution can also be obtained in slab-SDS-PAGE through a change in the conventional buffer system to tricine (58), or the use of polyacrylamide concentration gradient gels (59). Therefore, there are very effective methods to dissociate LPS mixtures in their monomers and separate, in a reproducible way, LPS native species with a high resolution through slab-PAGE.
However, it is important to notice that slab-PAGE technique had not been used in an extended form with preparative purposes until recent times because the most conventional and sensible of all methods for visualizing separate in gel LPS silver staining (55) originates the chemical modification and irreversible fixation of these macromolecules into the gel. This has conspired against individual LPS glycoforms recovery. As a consequence, localization and scission of LPS bands have had to be done in a very imprecise manner by sectioning unstained gels in two or three LPS band regions and its further comparison with other silverstained gels run in parallel. In general, the result continued showing much heterogeneity in the recuperated LPS fractions of slab-PAGE (42, 60, 61).
Preparative LPS separation by slab-PAGE is difficult due to: i) a high number of fractions should be recuperated from the gel (e.g., generally more than 10 for smooth type LPS); ii) high precision is needed to locate and divide the separated LPS bands, so as to avoid LPS fractions mixture that were previously resolved in the gel; iii) High LPS recuperation efficiency is demanded allowing to obtain enough quantities of minoritary LPS species for its evaluation in standard biological assays; iv) LPS should be recuperated from the gel without modifying their biological properties; and v) recuperated LPS from the gel should be free from detergents or salts associated to electrophoresis or elution; avoiding these chemicals could interfere in further LPS biological analyses.
Recently, at first stage, to overcome most obstacles, a sensitive and non-destructive reverse staining method of the LPS separated in gel was researched and established (43, 62, 63). At the second stage, it was known that individual LPS bands could diffuse in a fast and efficient way in a 5% triethylamine solution when extruded until producing gel microparticles of an average size 32 mm (64). Based on such principles, a new isolation methodology was established combining the separation in slab gel polyacrylamide, reverse staining, extrusion, and passive elution, to isolate to electrophoretic homogeneity microgram amounts of LPS individual species, starting from complex LPS mixtures of smooth or rough types (Figure 3) (65-67).
The amounts of obtained fractions from the two LPS sources (Vibrio fischeri HMK strain, and H.influenzae Rd) were enough for its structural analysis by MS either ESI or MALDI-TOF types (65, 66). For the first time, it was possible to demonstrate the relative abundance and structure characteristics of molecular species contained in each slab-PAGE band (e.g., the exact molecular mass, the oligosaccharidic sequence by ESI-MS or ESI-MS/MS, and lipid A phosphorylation status, the exact molecular mass of their intact oligosaccharides by ESI-MS or ESI-MS/MS or their O-desacylated by MALDITOF-MS). These analyses confirmed that the isolation method did not alter LPS chemical integrity (65, 66). This is the first time that separation with high resolution of LPS by slab- PAGE corresponded with the LPS structural analysis carried out by MS, which represents a significant improvement towards the direct correlation of chemical and biological assays.
This isolation method does not modify biochemical properties (e.g., TNF-α induction) of these biomolecules (63). Moreover, purified quantities of fifteen smooth type LPS individual species of E. coli K-235 (from 280 ng and 411 ng) were enough to quantify its activity at the LAL assay (67). For the first time, the LAL activity from smooth type LPS species was possible to be determined with a high level of electrophoretic homogeneity. Now that LPS molecules, in the amounts obtained by this methodology, are powerful inducers of diverse biological responses (e.g., induction of cells to produce TNF-α or nitric oxide in vitro, or lethal toxicity in mice sensitized with D-galactosamine in vivo), we look forward to using this method also for the analyses of LPS individual species in other biological assays.
This methodology owns several advantages compared to other methods for the separation of complex LPS molecular species. For the first time, the joint of smooth type LPS individual bands resolved by electrophoresis in a slab-polyacrylamide gel in presence of DOC (DOC-slab-PAGE) have been isolated up to electrophoretic homogeneity (67). Prior than this, the pattern of separated bands by electrophoresis in slab-polyacrylamide gel had been fractioned through scission in two or three regions from an unstained gel (59, 60). Though this method proved to be useful to separate rough type LPS fractions from smooth type LPS, it did not allow the precise fractioning of individual LPS bands. Logically, at the time of reanalyzing purified LPS fractions through electrophoresis, several species appeared (59, 60). Compared to the preparative DOC-PAGE method of cylindrical format, the isolation methodology based on separation by electrophoresis in smooth gel polyacrylamide also owns the advantage of providing a more precise detection and isolation of LPS individual species (51, 52). Besides, differently from the DOC-PAGE technique in cylindrical format, salts and detergents associated to electrophoresis are washed in the gel matrix before LPS elutes, and recovered fractions are released from these contaminants. The separation of LPS fractions based on DOC-lectrophoresis in slab-polyacrylamide gels offer several advantages upon other chromatographic methods which have been used to separate the whole LPS pattern (36-42, 68). These include a higher resolution, combined with a higher reproducibility at lower costs.
Although fractions obtained through the methodology based on electrophoresis in slab-polyacrylamide gel show a high electrophoretic homogeneity, being completely homogeneous in terms of molecular species is not expected (55, 65, 66). The analysis by MS of individual LPS fractions allowed detecting micro-heterogeneities that are below slab-PAGE resolution (65, 66). Mass spectra of the fraction with a lower LPS molecular mass of V. fischeri HMK, and the fractions of lower and higher molecular mass of H. influenzae LPS Rd, showed various molecular species (65, 66). At the time of correlating in an indirect way LPS band patterns generated by slab-polyacrylamide gel electrophoresis with mass spectra from LPS species (24, 69-71), it was confirmed that slab-PAGE is not, in an ordinary form, capable of solving small structural differences among the LPS species, like the presence of absence of low molecular weight replacement groups such as residues of ethanolamine, phosphates, or acetyl groups bound to the poly oligosaccharide skeleton or lipid A, nor LPS of close molecular masses resulting from the addition of a fatty acid to lipid A can be resolved. So, the next step towards the isolation of LPS molecular species up to chemical homogeneity level from complex mixtures of these glycolipids would probably need of other orthogonal separation principles (e.g., ionic exchange chromatography and RP-HPLC) in combination with slabpolyacrylamide gel electrophoresis.
Nevertheless, the exhaustive application of the isolation methodology of LPS individual species based on slab-polyacrylamide gel electrophoresis, combined with the simultaneous analysis of individual fractions by MS and biological assays may bring a new form of obtaining, in a more precise manner, information on chemical and functional heterogeneity, and the structure-function relations of LPS complex mixtures. These studies will have an important impact in the comprehension of LPS biosynthesis process and the role of these biomolecules in the physiopathology of bacterial infection, sepsis, and septic shock.
CONCLUSIONS
High structural heterogeneity of LPS and its amphipathic character makes the isolation of its molecular species extremely complex. Various methods have been applied for the purification of lipopolysaccharides with unsatisfactory results; yet in some of the following evaluation parameters: i) chemical homogeneity of LPS preparations; ii) preservation of the molecules chemical integrity; iii) applicability to diverse LPS sources; and iv) compatibility with structural and biological LPS characterization methods. LPS separation in a single chromatographic step has not led to the obtainment of LPS fractions of an adequate homogeneity. Even in its combined form, ionic exchange chromatographies and reversed phase, high resolution have allowed purification up to chemical homogeneity just from the simplest rough type LPS.
With the electrophoresis methods, including that of capillary format, preparative of cylindrical format or the slab-polyacrylamide, complex smooth type LPS have been separated. In this field, a new methodology combining electrophoresis techniques in slab-polyacrylamide, reverse staining, extrusion and passive elution have allowed the isolation of rough or smooth type intact LPS up to electrophoretic homogeneity. With this methodology, a sensible structural analysis by MS and the measurement of the biological activity of individual LPS species (intact) have been reached. It has been demonstrated that isolated fractions posses very few chemical species, and therefore, they are far more homogeneous than their original counterparts. The combination of electrophoresis with other orthogonal separation principles could probably constitute a next step forward towards the obtainment of chemically homogeneous preparations starting from complex LPS mixtures.
REFERENCES
1. Morrison DC, Silverstein R, Luchi M, Shnyra A. Structure-function relationships of bacterial endotoxins. Contribution to microbial sepsis. Infect Dis Clin North Am 1999;13:313-40.
2. Rietschel ET, Brade H, Holst O, Brade L, Muller-Loennies S, Mamat U, et al. Bacterial endotoxin: Chemical constitution, biological recognition, host response, and immunological detoxification. Curr Top Microbiol Immunol 1996;216:39-81.
3. Pier GB. Promises and pitfalls of Pseudomonas aeruginosa lipopolysaccharide as a vaccine antigen. Carbohydr Res 2003;338:2549-56.
4. Hu WG, Berry J, Chen J. Exploration of Moraxella catarrhalis outer membrane proteins, CD and UspA, as new carriers for lipooligosaccharide-based conjugates. FEMS Immunol Med Microbiol 2004;41:109-15.
5. McCluskie MJ, Weeratna RD. Novel adjuvant systems. Curr Drug Targets Infect Disord 2001;1:263-71.
6. Caroff M, Karibian D. Structure of bacterial lipopolysaccharides. Carbohydr Res 2003;338:2431-47.
7. Muroi M, Tanamoto KI. The polysaccharide portion plays an indispensable role in Salmonella lipopolysaccharide-induced activation of NF-kappaB through human toll-like receptor 4. Infect Immun 2002;70:6043-7.
8. Brade L, Engel R, Christ WJ, Rietschel ETh. A non-substituted primary hydroxyl group in position 6' of free lipid A is required for binding of lipid A monoclonal antibodies. Infect Immun 1997;65:3961-5.
9. Caroff M, Cavaillon JM, Fitting C, Haeffner-Cavaillon N. Inability of pyrogenic, purified Bordetella pertussis lipid A to induce interleukin-1 release by human monocytes. Infect Immun 1986;54:465-71.
10. Haeffner-Cavaillon N, Caroff M, Cavaillon JM. Interleukin-1 induction by lipopolysaccharides: structural requirements of the 3-deoxy-D-manno-2-octulosonic acid (KDO). Molec Immunol 1989;26:485-94.
11. Haeffner-Cavaillon N, Carreno MP, Aussel L, Caroff M. Molecular aspects of endotoxins relevant to their biological functions. Nephrol Dial Transplant 1999;14:853-60.
12. Zahringer U, Lindner B, Rietschel ET. Chemical structure of lipid A. Recent advances in structural analysis of biologically active molecules . In: Brade H, Opal SM, Vogel SN, Morrison DC (editors). Endotoxin in Health and Disease. New York: Marcel Dekker Inc;1999, p. 3-114.
13. Laude-Sharp M, Haeffner-Cavaillon N, Caroff M, Lantreibecq F, Pusineri C, Kazatchkine MD. Dissociation between the interleukin 1-inducing capacity and Limulus reactivity of lipopolysaccharides from gram-negative bacteria. Cytokine 1990;2:253-8.
14. Koyama S, Sato E, Nomura H, Kubo K, Miura M, Yamashita T, et al. The potencial of various lipopolysaccharides to release IL-8 and G-CSF. Am J Physiol 2000;278:L658-66.
15. Paeng N, Kido N, Schmidt G, Sugiyama T, Kato Y, Koide N, et al. Augmented immunological activities of recombinant lipopolysaccharide possessing the mannose homopolymer as the O-specific polysaccharide. Infect Immun 1996;64:305-9.
16. Blondiau C, Lagadec P, Lejeune P, Onier N, Cavaillon JM, Jeannin JF. Correlation between the capacity to activate macrophages in vitro and the antitumor activity in vivo of lipopolysaccharides from different bacterial species. Immunobiology 1994;190:243-54.
17. Kirikae T, Schade FU, Kirikae F, Rietschel ETh, Morrison DC. Isolation of a macrophage-like cell line defective in binding of lipopolysaccharide. Influence of serum and lipopolysaccharide chain length on macrophage activation. J Immunol 1993;151:2742-52.
18. Freudenberg MA, Freudenberg N, Galanos C. Time course of cellular distribution of endotoxin in liver, lungs and kidneys of rats. Br J Exp Path 1982;63:56-65.
19. Freudenberg MA, Bog-Hansen TC, Back U, Galanos C. Interaction of lipopolysaccharides with plasma high-density lipoprotein in rats. Infect Immun 1980;28:373-80.
20. Huber M, Kalis C, Keck S, Jiang Z, Georgel P, Du X, et al. R-form LPS, the master key to the activation ofTLR4/MD-2-positive cells. Eur J Immunol 2006;36:701-11.
21. Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem 2002;71:635-700.
22. Lamari FN, Gioldassi XM, Mitropoulou TN, Karamanos NK. Structure analysis of lipoglycans and lipoglycan-derived carbohydrates by capillary electrophoresis and mass spectrometry. Biomed Chromatogr 2002;16:116-26.
23. Li J, Thibault P, Martin A, Richards JC, Wakarchuk WW, Van der Wilp W. Development of an on-line preconcentration method for the analysis of pathogenic lipopolysaccharides using capillary electrophoresis-electrospray mass spectrometry. Application to small colony isolates. J Chromatogr A 1998;817:325-36.
24. Caroff M, Deprun C, Karibian D. 252Cf plasma desorption mass spectrometry applied to the analysis of underivatized rough-type endotoxin preparations. J Biol Chem 1993;268:12321-4.
25. Czaja J, Jachymek W, Niedziela T, Lugowski C, Aldova E, Kenne L. Structural studies of the O-specific polysaccharide from Plesiomonas shigelloides strain CNCTC 113/92. Eur J Biochem 2000;267:1672-9.
26. Plotz BM, Lindner B, Stetter KO, Holst O. Characterization of a novel lipid A containing D-galacturonic acid that replaces phosphate residues. The structure of the lipid A of the lipopolysaccharide from the hyperthermophilic bacterium Aquifex pyrophilus. J Biol Chem 2000;275:11222-8.
27. Jiao BH, Freudenberg M, Galanos C. Characterization of the lipid A component of genuine smooth-form lipopolysaccharide. Eur J Biochem 1989;180:515-8.
28. Lebbar S, Karibian D, Deprun C, Caroff M. Distribution of lipid A species between long and short chain lipopolysaccharides isolated from Salmonella, Yersinia, and Escherichia as seen by 252Cf plasma desorption mass spectrometry. J Biol Chem 1994;269:31881-4.
29. Jarvis BW, Qureshi N. Inhibition of lipopolysaccharide-induced transcription factor Sp1 binding by spectrally pure diphosphoryl lipid A from Rhodobacter sphaeroides, protein kinase inhibitor H-8, and dexamethasone. Infect Immun 1997;65:1640-3.
30. Aurell CA, Wistrom AO. Critical aggregation concentrations of Gram negative bacterial lipopolysaccharides (LPS). Biochem Biophys Res Commun 1998;253:119-23.
31. Ribi E, Anacker R, Brown W, Haskins WT, Malmgren B, Milner KC, et al. Reaction of endotoxin and surfactants. I. Physical and biological properties of endotoxin treated with sodium deoxycholate. J Bacteriol 1966;92:1493-1509.
32. Shands JW Jr, Chun PW. The dispersion of gram-negative lipopolysaccharide by deoxycholate. Subunit molecular weight. J Biol Chem 1980;255:1221-6.
33. Westphal O, Jann K. Bacterial lipopolysaccharides: extraction with phenol-water and further applications of the procedure. Methods Carbohydr Chem 1965;5:83-91.
34. Galanos C, Luderitz O, Westphal O. A new method for the extraction of R lipopolysaccharides. Eur J Biochem 1969;9:245-9.
35. Hitchcock PJ, Brown TM. Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J Bacteriol 1983;154:269-77.
36. Suda Y, Shiyama T, Yasukochi T, Kawano K, Brade H, Rietschel ET, et al. Separation of wild-type lipopolysaccharides by centrifugal partition chromatography: isolation of smooth form species. J Endotoxin Res 1996;3:1-7.
37. Yeh HY, Jacobs DM. Characterization of lipopolysaccharide fractions and their interactions with cells and model membranes. J Bacteriol 1992;174:336-41.
38. Nowotny A. Heterogeneity of endotoxic bacterial lipopolysaccharides revealed by ion-exchange column chromatography. Nature 1966;210:278-80.
39. Fischer W. Purification and fractionation of lipopolysaccharide from gram-negative bacteria by hydrophobic interaction chromatography. Eur J Biochem 1990;194:655-61.
40. Le Dur A, Chaby R, Szabo L. Isolation of two protein-free and chemically different lipopolysaccharides from Bordetella pertussis phenol-extracted endotoxin. J Bacteriol 1980;143:78-88.
41. Kol O, Montreuil J, Fournet B, Zalisz R, Smets P. Purification of the lipopolysaccharide fraction from Klebsiella pneumonia O1 K2 by high-performance liquid chromatography. J Chromatogr 1987;396:281-6.
42. Komuro T, Yomota C, Kimura T, Galanos C. Comparison of R- and S-form lipopolysaccharides fractionated from Escherichia coli UKT-B lipopolysaccharide in pyrogen and Limulus tests. FEMS Microbiol Lett 1989;51:79-83.
43. Hardy E, Pupo E, Castellanos-Serra L, Reyes J, Fernández-Patrón C. Sensitive reverse staining of bacterial lipopolysaccharides on polyacrylamide gels by using zinc and imidazole salts. Anal Biochem 1997;244:28-32.
44. Therisod H, Labas V, Caroff M. Direct microextraction and analysis of roughtype lipopolysaccharides by combined thin-layer chromatography and MALDI mass spectrometry. Anal Chem 2001;73:3804-7.
45. Chen CH, Chang C, Nowotny AM, Nowotny A. Rapid biological and chemical analyses of bacterial endotoxins separated by preparative thin-layer chromatography. Anal Biochem 1975;63:183-94.
46. Qureshi N, Takayama K, Mascagni P, Honovich J, Wong R, Cotter RJ. Complete structural determination of lipopolysaccharide obtained from deep rough mutant of Escherichia coli. Purification by high performance liquid chromatography and direct analysis by plasma desorption mass spectrometry. J Biol Chem 1988;263:11971-6.
47. Zamfir A, Peter-Katalinic J. Capillary electrophoresis-mass spectrometry for glycoscreening in biomedical research. Electrophoresis 2004;25:1949-63.
48. Kelly J, Masoud H, Perry MB, Richards JC, Thibault P. Separation and characterization of O-deacylated lipo-oligosaccharides and glycans derived from Moraxella catarrhalis using capillary electrophoresis-electrospray mass spectrometry and tandem mass spectrometry. Anal Biochem 1996;233:15-30.
49. Thibault JL, Martin A, Richards JC, Wakarchuk WW, van der Wilp W. Development of an on-line preconcentration method for the analysis of pathogenic lipopolysaccharides using capillary electrophoresis-electrospray mass spectrometry. Application to small colony isolates. J Chromatogr A 1998;817:325-36.
50. Horton D, Riley DA. 31P nuclear magnetic resonance spectroscopy of lipopolysaccharides from Pseudomonas aeruginosa. Biochim Biophys Acta 1981;640:727-33.
51. Knirel YA, Helbig JH, Zahringer U. Structure of a decasaccharide isolated by mild acid degradation and dephosphorylation of the lipopolysaccharide of Pseudomonas fluorescens strain ATCC 49271. Carbohydr Res 1996;283:129-39.
52. Volpi N. Separation of Escherichia coli 055:B5 lipopolysaccharide and detoxified lipopolysaccharide by high-performance capillary electrophoresis. Electrophoresis 2003;24:3097-103.
53. Kim JS, Reuhs BL, Rahman MM, Ridley B, Carlson RW. Separation of bacterial capsular and lipopolysaccharides by preparative electrophoresis. Glycobiology 1996;6:433-7.
54. Rahman MM, Guard-Petter J, Carlson RW. A virulent isolate of Salmonella enteritidis produces a Salmonella typhi-like lipopolysaccharide. J Bacteriol 1997;179:2126-31.
55. Tsai CM, Frasch CE. A sensitive silver stain for detecting lipopolysaccharides in polyacrylamide gels. Anal Biochem 1982;5:115-9.
56. Peterson AA, McGroarty EJ. High molecular-weight components in lipopolysaccharides of Salmonella typhimurium, Salmonella minnesota, and Escherichia coli. J Bacteriol 1985;162:738-45.
57. Komuro T, Galanos C. Analysis of Salmonella lipopolysaccharides by sodium deoxycholate-polyacrylamide gel electrophoresis. J Chromatogr 1988;450:381-7.
58. Lesse AJ, Campagnari AA, Bittner WE, Apicella MA. Increased resolution of lipopolysaccharides and lipopoligosaccharides utilizing tricine-sodium dodecyl sulfate polyacrylamide gel electrophoresis. J Immunol Methods 1990;126:109-17.
59. Noda K, Kubota K, Yamasaki R. Separation of lipooligosaccharides by linear gradient gel electrophoresis. Anal Biochem 2000;279:18-22.
60. DiRienzo JM, MacLeod RA. Composition of the fractions separated by polyacrylamide gel electrophoresis of the lipopolysaccharide of a marine bacterium. J Bacteriol 1978;136:158-67.
61. Ohta M, Rothmann J, Kovats E, Phan PH, Nowotny A. Biological activities of lipopolysaccharide fractionated by preparative acrylamide gel electrophoresis. Microbiol Immunol 1985;29:1-12.
62. Hardy E. Detección con zinc-imidazol de biomoléculas en geles: aplicaciones analíticas y micropreparativas [ dissertation]. La Habana: Habana Univ.;1998 .
63. Hardy E, Pupo E, Santana H, Guerra M, Castellanos-Serra L. Elution of lipopolysaccharides from polyacrylamide gels. Anal Biochem 1998;259:162-5.
64. Pupo E, López CM, Alonso M, Hardy E. High-efficiency passive elution of bacterial lipopolysaccharides from polyacrylamide gels. Electrophoresis 2000;21:526-30.
65. Pupo E, Phillips NJ, Gibson BW, Apicella MA, Hardy E. Matrix-assisted laser desorption/ionization-time of flight-mass spectrometry of lipopolysaccharide species separated by slab-polyacrylamide gel electrophoresis: high resolution separation and molecular weight determination of lipooligosaccharides from Vibrio fischeri strain HMK. Electrophoresis 2004;25:2156-64.
66. Gulin S, Pupo E, Schweda EK, Hardy E. Linking mass spectrometry and slab-polyacrylamide gel electrophoresis by passive elution of lipopolysaccharides from reverse-stained gels: analysis of gel-purified lipopolysaccharides from Haemophilus influenza strain Rd. Anal Chem 2003;75:4918-24.
67. Pupo E, Hardy E. Isolation of smoothtype lipopolysaccharides to electrophoretic homogeneity. Electrophoresis 2007;28:2351-7.
68. Goodman MG, Morrison DC, Weigle WO. Modulation of lipopolysaccharide (LPS)-mediated function by structural differences of two physically distinct fractions of Escherichia coli K235 LPS. J Immunol 1977;118:1852-7.
69. Gibson BW, Campagnari AA, Melaugh W, Phillips NJ, Apicella MA, Grass S, et al. Characterization of a transposon Tn916-generated mutant of Haemophilus ducreyi 35000 defective in lipooligosaccharide biosynthesis. J Bacteriol 1997;179:5062-71.
70. Cox AD, Hood DW, Martin A, Makepeace KM, Deadman ME, Li J, et al. Identificationand structural characterization of a sialylated lacto-N-neotetraose structure in the lipopolysaccharide of Haemophilus influenzae. Eur J Biochem 2002;269:4009-19.
71. Post DM, Phillips NJ, Shao JQ, Entz DD, Gibson BW, Apicella MA. Intracellular survival of Neisseria gonorrhoeae in male urethral epithelial cells: importance of a hexaacyl lipid A. Infect Immun 2002;70:909-20.
Received inDecember 2007.
Accepted for publication inJuly 2008.
Elder Pupo. Laboratorio de Polímeros, Instituto de Ciencia y Tecnología de los Materiales, Universidad de La Habana, La Habana, Cuba. Correo electrónico: ehardy@imre.oc.uh.cu

{kind=link}
{kind=link}