










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBiotecnología Aplicada
versión On-line ISSN 1027-2852
Biotecnol Apl v.26 n.2 La Habana abr.-jun. 2009
REVIEW
Methodology for guidelines development for biopharmaceuticals obtained from transgenic plants in Cuba
Metodología para la elaboración de las pautas reguladoras para productos biofarmacéuticos obtenidos de plantas transgénicas en Cuba
Yanet Hechavarría-Nuñez, Lázara Martínez-Muñoz, Rodrigo O Pérez-Massipe, Yohanka Martínez-Gzegozewska, Olga L Jacobo-Casanueva, Rolando B Domínguez-Morales
Centro para el Control Estatal de la Calidad de los Medicamentos, CECMED Calle 200 No. 1706 e/ 17 y 19, Reparto Siboney, CP 11600, AP 16065, Ciudad de La Habana, Cuba
ABSTRACT
Molecular farming is an evolving field in Cuba; therefore, establishing the guidelines guaranteeing the quality, safety and efficacy of biopharmaceutical products is a challenge for the National Regulatory Agency (Center for State Control on the Quality of Drugs; CECMED). The methodology to elaborate guidelines for this type of products was mainly based on the revision of the legal and regulatory international framework, the evaluation of the current and prospective developmental stage of the technology in Cuba and the comparative analysis among the manufacturing process steps of a biotechnological product obtained in traditional expression systems and that derived from transgenic plants. A first draft of the regulatory document was compared to guidance drafts issued by other regulatory agencies (such as FDA and EMEA) and extensively reviewed by CECMED specialists as well as by experts of the local biotech industry. As a result, a regulation project was elaborated, and its applicability tested while the assessment of an application for scientific advisory, related to product obtained by this technology. Finally, the regulation was approved, being applicable for products generated in genetically modified plants. It is also valid for edible vaccines and other compounds which do not constitute active pharmaceutical ingredients.
Keywords: transgenic plants, biopharmaceuticals, guidance, regulatory guidelines, CECMED.
RESUMEN
La obtención de productos biofarmacéuticos a partir de plantas transgénicas es un campo en evolución en Cuba, por lo que la elaboración de pautas reguladoras que garanticen su calidad, seguridad y eficacia constituye un reto para la Autoridad Reguladora de Medicamentos, el Centro para el Control Estatal de la Calidad de los Medicamentos, CECMED. La metodología para establecer las pautas reguladoras para estos productos se basó fundamentalmente en la revisión del entorno legal y regulador internacional, la evaluación del estado de desarrollo actual y prospectivo de la tecnología en Cuba, y en la comparación de las diferentes etapas del proceso de obtención de un producto biotecnológico en sistemas tradicionales de expresión y a partir de plantas transgénicas. La primera versión del documento normativo se comparó con las guías preliminares emitidas por otras autoridades reguladoras, como la Administración de Alimentos y Medicamentos (FDA) y la Agencia Europea para la Evaluación de Medicamentos (EMEA) y la revisaron especialistas del CECMED y de la industria. Como resultado, se elaboró un proyecto de regulación, cuya aplicabilidad se comprobó mediante la evaluación de un trámite de registro de un producto cubano obtenido utilizando esta tecnología. Finalmente, se aprobó un documento normativo aplicable a proteínas obtenidas de plantas genéticamente modificadas, válido también para la obtención de vacunas comestibles y de otros compuestos que no constituyan el principio activo.
Palabras clave: plantas transgénicas, productos biofarmacéuticos, documento regulador, pautas reguladoras, CECMED.
INTRODUCTION
The use of genetically modified crops to obtained pharmaceutically active molecules increased over the last years. Among their multiple advantages to produce active pharmaceutical ingredients are the production of a wide range of proteins and the considerable reduction of investment-operational costs, while minimizing contamination risks with pathogens harmful to mankind (1).
As a system designed to produce pharmaceutically active molecules, the process to obtain proteins in transgenic plants should comply with regulatory guidelines to guarantee quality, safety and efficacy of the product. Some of the regulations formerly established for pharmaceutical production process in microorganisms and mammalian cells could be applied to some steps of this novel technology. However, other production steps are unattended, due to the properties of the host system employed (2).
Research projects are under development to produce biopharmaceutical products in transgenic plants in Cuba. Thus, the elaboration of regulatory guidelines has become a challenge for the National Regulatory Agency (NRA), because there are no harmonized regulations internationally available in this field. For this reason, the Center for State Control on the Quality of Drugs (CECMED), the Cuban National Regulatory Agency, has established the main aspects to be considered for documenting the approval of biopharmaceutical products obtained in transgenic plants. These aspects are in agreement with the development achieved by the Cuban biotech-pharmaceutical industry.
IDENTIFYING THE NATIONAL AND INTERNATIONAL FRAMEWORK
A precise regulatory environment is required to pharmacologically active biomolecules, obtained in plants by using them as bioreactors in order to protect human health. Only FDA and EMEA have issued regulatory documents on this type of productions (3, 4), which are currently reviewed. The World Health Organization held an informal consultation on regulatory evaluation of candidate human vaccines from plants, in January 2005, for industry and NRAs experts. As a result, a report was issued summarizing the topics discussed and conclusions include the points to be considered to elaborate a regulatory document in this field (5).
For the present work, it was necessary to identify the Cuban scientific institutions involved at the different phases of the research & development process of genetically modified plants (Table 1). The Centers for Genetic Engineering and Biotechnology (CIGB) of Havana, Santi Spiritus and Camagüey, the Center for Bioplants at the University of Ciego de Avila and the Plant Biotechnology Institute of Villa Clara constitute the leading front for the development of this technology in Cuba. Other institutions related to the evaluation and field introduction of transgenic plants as collateral activities, depending on their missions, are summarized in table 1. This also evidences the tight collaboration among the Cuban scientific institutions.

The working strategies are mainly directed to obtain economically relevant crops (sweet potato, pineapple, sugar cane, potato, tomato, corn, papaya, coffee, citrus, rice and banana) resistant to herbicides and pathogens (6). Only the CIGB of Havana and Santi Spiritus have research projects on the expression of pharmacologically active proteins in transgenic plants to be used as active ingredients or biological reagents in the biopharmaceutical industry. For example, the CIGB of Havana produces a monoclonal antibody (MAb) in tobacco (Nicotiana tabacum) transgenic plants (7). This MAb is employed for the purification of the active pharmaceutical ingredient (API) of the Heberbiovac HB® recombinant anti-hepatitis B vaccine.
Besides, the scope of the National Program for Biotechnology of Agriculture and Livestock (8) comprises the production of drugs and oral vaccines for animals and humans through efficient genetic transformation methods.
The National Center for Biological Safety (the regulatory authority for biosafety) has established legal dispositions ruling the use, management, storage, transport, import and export of genetically-modified organisms (9-12). However, there is an urgent need for specific regulations guaranteeing the quality, safety and efficacy of biopharmaceutical products obtained in transgenic plants for human use.
ELABORATION OF THE DRAFT GUIDANCE
A definition of the legal category was required to design the draft regulating the steps for the approval of biopharmaceutical products obtained in transgenic plants. First, the national requirements established for marketing authorization of pharmaceutical products, Requisitos para las solicitudes de inscripción, renovación y modificación del registro de medicamentos de uso humano (13), were analyzed, particularly chapters related to products obtained by the recombinant DNA technology. We concluded that these requirements did not cover the regulation of transgenic plant biotechnological products, particularly because of not dealing with the cellular substrates used as starting materials in the process. Moreover, the need for an introductory chapter bringing general information for the applicants was evident, as well as a chapter of definitions for specific terms of this technology and other chapters regarding the regulatory claims. Consequently, and considering what established in the institutional documentation of the CECMED, the legal document should be adopted in the format of a regulation ruling the formalities for marketing authorization of biopharmaceutical products obtained in transgenic plants. This document will complement marketing authorization requirements in force.
The document was elaborated following what established in the standard operation procedure (SOP): Metodología para la elaboración, aprobación y revision de regulaciones of the CECMED (14); the different chapters of regulatory contents were defined from the analyses of the regulatory guidelines on the Common Technical Document (15); but the order of the sections described in the Quality Module III was adapted to comply with the Cuban marketing authorization requirements in force, accordingly.
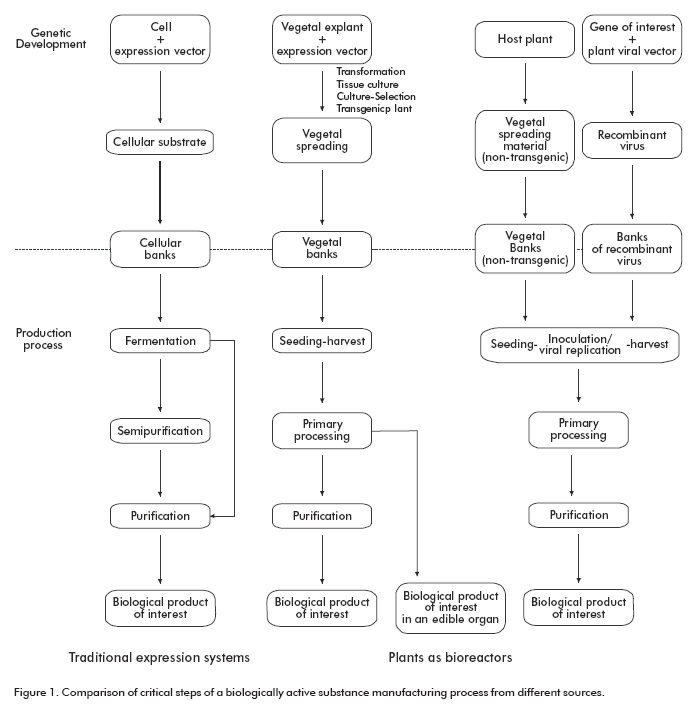
The regulatory requirements were established on the basis of identifying critical steps, by comparing the diagrams of recombinant DNA products manufacturing process in transgenic plants and in traditional host systems (microorganisms and eukaryotic cells) (Figure 1). The critical steps to be controlled within predetermined criteria, to guarantee an API complying with quality specifications (16), were identified taking as standards the critical steps established in traditional host system-based process for bipharmaceutical production. Similarities between the purposes for each step were also considered. Besides, parameters to be monitored during the entire process were identified, and other variants of this technology were analyzed, such as the use of viral vectors.
Due to the lack of specific international regulations for this technology at the beginning of this investigation, a vast regulatory documentation was consulted during the drafting phase of the document, such as regulations on the process to obtain biologicals/biotechnological products from recombinant microorganisms and eukaryotic cells (17-21); regulations concerning natural products, because this technology shares an agricultural phase (22, 23); regulations for the environmental protection and the introduction of genetically modified organisms (24), and others established for transgenic foods (25), because of the possibility to use vegetables or fruits as drug delivery systems (e.g. edible vaccines).
The proposed guideline was compared to the draft guidance issued by the FDA (3) and EMEA (26), to harmonize criteria and considering their experience on this topic, due to participation on scientific debates and also due to the majority of the research projects are being carried out in developed countries. Additionally, we examined the comments of the FDA draft guidance expressed by the Biotechnology Industry Organization on behalf of the industrial sector (27), and also those from the Monsanto (28), ProdiGene (29) and Meristem Therapeutics (30) companies, respectively. The draft was submitted to an internal evaluation process by CECMED specialists, to analyze and determine if the information requested was enough or if additional data were required, also contributing to the correct style of the document. Subsequently, the document was completed and subjected to external evaluation by researchers, production specialists and the Regulatory Affairs group and management staff of the Agriculture and Livestock Division at the CIGB of Havana. Once received the comments, the regulation project was ready to be prepared.
PREPARATION OF GUIDANCE PROJECT
A debate with the Industry allowed the use of harmonized terms and regulatory language in the guidelines, in agreement with the national situation and international tendencies. Most relevant observations concerned the scope of the document, i.e., if the document was applicable for molecules used as reagents, the need for a section about edible vaccines and the inclusion of new and more appropriate terms and definitions. Other comments were about the number of lots required for consistency demonstration and about parameters to characterize vegetal banks, particularly the bioburden.
The project was structured accordingly, comprising the possible methods to obtain biopharmaceuticals in genetically modified plants and including additional information and definitions.
The guideline drafted was put to the consideration of specialists from the CECMED and other institutions such as the CIGB of Havana, the Institute for Food Hygiene and Health, the Ministry of Agriculture and the National Center for Biological Safety.
It was also simultaneously challenged against through the assessment of an application for scientific advisory.
PRELIMINARY EVALUATION OF GUIDANCE PROJECTS IMPACT
The project was finally applied to a scientific advisory dossier for the production of a monoclonal antibody in Nicotiana tabacum transgenic plants. This monoclonal antibody is used to purify the hepatitis B surface antigen.
As a result of this practical exercise, it was concluded that the document complies with the regulatory purposes it was intended for, involving the main aspects of production and control of a product obtained in transgenic plants. During scientific meetings with the applicant, it was demonstrated that the document constitutes an agreed regulating solution to the national scenario.
PREPARATION OF THE FINAL REGULATORY DOCUMENT
The regulatory document was elaborated considering results from previous stage, external opinions as well as the state on this subject at the time of writing the document. The structure of the final document is shown in table 2.

The document includes among regulatory aspects those related to the main concerns identified during the revision of the national and international framework (31-37), which are related as follows:
a) The existence of two different ways for the expression of heterologous proteins in plants: stable integration of the transgene(s) into the plant´s genome, and the transient expression by using plant viral vectors.
b) The need of establishing a banking system from structures of easy reproduction and storage to guarantee production consistency.
c) Factors influencing plant´s growth and development, such as soil, irrigation, fertilization, incidence of pests, sunlight and other climatic factors.
d) Measures to avoid the flux of pollen and seeds between plants and the environment.
e) Measures to minimize fungal and bacterial contamination levels.
f) Procedures to obtain the compound of interest: extraction and purification from the harvested vegetal material or its expression in plant edible organs.
g) Possible functional and structural differences in the protein obtained, in respect to its native conformation, especially for glycosylated proteins.
h) The presence of contaminants from the host plant (proteins, DNA, alkaloids), the culture media (heavy metals, pesticides traces, fertilizers) and productassociated impurities (aggregates, truncated and modified molecular species).
i) Expression levels variability and compliance with quality specifications among production lots.
j) Presence of new toxic compounds or their increase, or higher anti-nutrients levels in plant edible organs.
The regulatory document was approved on April 2006 (38) and used in the assessment of a marketing authorization application related to the alternate use of a Mab produced in Nicotiana transgenic plants, for the purification of the Hepatitis B surface antigen (39-41).
In summary, a regulatory document, applicable to biopharmaceutical products obtained in transgenic plants is available at the CECMED to fulfill our national regulatory needs. This document can be used as reference to design research & development projects, also including regulatory requirements since the beginning of research to guarantee the proper development of new products.
Noteworthy, because of the novelty of this technology and its incipient exploitation by Cuban institutions, these guidelines can be modified as part of the updating process according to the state of the art. This seems to be the most common scenario faced by the NRAs throughout the world (41, 42), since every host plant system and biological product becomes a new challenge due to their unique properties. Collaboration between the industry, the regulatory agencies and the specialists involved is the only way to solve the evolving problems of technological development.
REFERENCES
1. Twyman RM, Schillberg S, Fisher R. Transgenic plants in the biopharmaceutical market. Expert Op Emerg Drugs 2005;10(1):185-218.
2. CPMP/BWP/1711/00. Concept paper on the development of a Committee for Propietary Medicinal Products (CPMP) points to consider on the use of transgenic plants in the manufacture of biological medicinal products for human use.2001 march. (En línea) Disponible en: http://www.emea.europa.eu/pdfs/human/bwp171100en.pdf
3. Food and Drugs Administration (FDA). Guidance for Industry Drugs, Biologics and Medical Devices Derived from Bioengineered Plants for Use in Humans and Animals. Draft guidance 2002) sept. (En línea) Disponible en: www.fda.gov/cber/guidelines.htm.
4. EMEA/CHMP/BWP/48316/2006. Guideline on the quality of biological active substances produced by stable transgene expression in higher plants. Draft guidance-2 2006) jul. (En línea) Disponible en: http://www.emea.europa.eu/htms/human/humanguidelinesbiologicals.htm.
5. World Health Organization. (WHO). Report: WHO informal consultation on scientific basis for regulatory evaluation of candidate human vaccines from plants 2005 jan. (En línea) Disponible en: http://www.who.int/biologicalspublications/meetings/areas/vaccines/plants/en/index.html.
6. Arrieta J, Torres V (editor-compilador). Biotecnología Habana 2005. Resúmenes. La Habana: Elfos Scientiae,2005.
7. Pujol M, Ramírez NI, Ayala M, Gavilondo JV, Valdés R, Rodríguez M, et al. An intregral approach towards a practical application for a plant-made monoclonal antibody in vaccine purification. Vaccine 2005;23:1333-7.
8. Borroto C, Pérez MC, Cornide MT, Peralta LE, Fernández-Larrea O, Fundora Z, et al. Programa Nacional de Biotecnología Agropecuaria. Programas Nacionales Científicos. (En línea) Disponible en: www.geprop.cu.
9. Centro Nacional de Seguridad Biológica (CNSB). Decreto ley 190 de la Seguridad Biológica. (En línea) Disponible en: www.medioambiente.cu/oregulatoria/cnsb/legislación/legislación1.htm.
10. Ministerio de Ciencia, Tecnología y Medioambiente (CITMA). Resolución 8/ 2000: Reglamento general de seguridad biológica para las instalaciones en las que se manipulan agentes biológicos y sus productos, organismos y fragmentos de estos con información genética.
11. Ministerio de Ciencia, Tecnología y Medioambiente (CITMA). Resolución 76/ 2000: Reglamento para el otorgamiento de las autorizaciones de seguridad biológica.
12. Ministerio de Ciencia, Tecnología y Medioambiente (CITMA). Resolución 103/ 2002: Reglamento para el establecimiento de los requisitos y procedimientos de seguridad biológica en las instalaciones en las que se hace uso de agentes biológicos y sus productos, organismos y fragmentos de estos con información genética.
13. Ministerio de Salud Pública (MINSAP). Resolución 168: Requisitos para las solicitudes de inscripción, renovación y modificación en el registro de medicamentos de uso humano (anexos 5 y 6) 2000 oct 4. (En línea) Disponible en: http://www.cecmed.sld.cu/Docs/RegFarm/DRA/Lic Prod/1992-2000/Reg/ReqSol_IRMR.pdf.
14. Centro para el Control Estatal de la Calidad de los Medicamentos (CECMED). PNO 07.001: Metodología para la elaboración, aprobación y revisión de regulaciones, 1ra ed.,1995 nov 29.
15. ICH. M4Q: The CTD-Quality Guidance for Industry 2001 aug. (En línea) Disponible en: http://www.ich.org/cache/compo/276-254-1.html.
16. ICH. Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients (Q7A) 2000. (En línea) Disponible en: http://www.ich.org/cache/compo/276-254-1.html.
17. ICH. Quality of Biotechnological Products: Analysis of the expression construct in cells used for production of rDNA derived products (Q5B) 1997. (En línea) Disponible en: http://www.ich.org/cache/compo/276-254-1.html.
18. ICH. Derivation and characterization of cell substrates used for production of biotechnological/biological products (Q5D) 1997. (En línea) Disponible en: http://www.ich.org/cache/compo/276-254-1.html.
19. ICH. Specifications: Test procedures and acceptance criteria for biotechnological / biological products (Q6B) 1999. (En línea) Disponible en: http://www.ich.org/cache/compo/276-254-1.html.
20. Organización Mundial de la Salud (OMS). Anexo 3: Pautas para el Control de la Calidad de los productos farmacéuticos y biológicos preparados mediante técnicas del ADNr. Serie de Informes Técnicos; 814. 1991.
21. Food and Drug Administration (FDA). Content and format of Chemistry, Manufacturing and Controls Information and Establisment Description Information for a Vaccine or Related Product. Guidance for Industry 1999). (En línea) Disponible en: www.fda.gov/cber/guidelines.htm.
22. EMEA/HMPWP/31/99 Rev.3. Points to Consider on Good Agricultural and Collection Practice for Starting Materials of Herbal Origin 2002 may. (En línea) Disponible en: www.emea.eu.int.
23. Centro para el Control Estatal de la Calidad de los Medicamentos (CECMED). Regulación 28/2002: Requisitos para las solicitudes de inscripción, renovación y modificación en el registro de medicamentos de origen natural de uso humano 2002 may. (En línea) Disponible en: http://www.cecmed.sld.cu/Docs/RegFarm/DRA/LicProd/2001-2002/Reg/Reg_28-02.pdf.
24. CFIA. Guidelines for the Enviromental Release of Plants with Novel Traits within Confined Field Trials in Canada 2000 jul. (En línea) Disponible en: www.inspection.gc.ca/english/plaveg/biol/dirdir0007e.shtml.
25. Health Protection Branch/ Food Directorate. Guideline for the safety assesment of novel foods. Volume II Genetically Modified Microorganisms and Plants 1994 sept. (En línea) Disponible en: http://www.hc-sc.gc.ca/ fn-an/legislation/guide-ld/nvvlii01_e.html.
26. CPMP/BWP/764/02. Points to Consider on Quality Aspects of Medicinal Products Containing Active Substances Produced by Stable Transgene Expression in Higher Plants. Draft guideline-12002 march. (En línea) Disponible en: www.emea.eu.int.
27. BIO. Comments in response to the Draft Guidance for Industry Drugs, Biologics and Medical Devices Derived from Bioengineered Plants for Use in Humans and Animals 2003 feb. (En línea) Disponible en: http://www.fda.gov/ohrms/dockets/dailys/03/Feb03021903/02d-0324-c000607-01-vol13.pdf.
28. Monsanto. Monsanto Company Response to Draft Guidance for Industry Drugs, Biologics and Medical Devices Derived from Bioengineered Plants for Use in Humans and Animals 2003 jan. (En línea) Disponible en: http://www.fda.gov/ohrms/dockets/dailys/03/Jan03/010703/800467db.pdf.
29. ProdiGene. Comments on the Draft Guidance for Industry Drugs, Biologics and Medical Devices Derived from Bioengineered plants for Use in Humans and Animals 2003 jan. (En línea) Disponible en: http://www.fda.gov/ohrms/dockets/dailys/03/Jan03/012103/8004a378.html.
30. Meristems Therapeutics. Comments on the Draft Guidance for Industry Drugs, Biologics and Medical Devices Derived from Bioengineered Plants for Use in Humans and Animals 2003 jan. (En línea) Disponible en: http://www.fda.gov/ohrms/dockets/dailys/03/Jan03/011603/8004ac98.pdf.
31. Miele L. Plants as biorreactors for biopharmaceuticals: regulatory considerations. Trends Biotechnol 1997;15(2):45-50.
32. Stein KE, Webber KO. The regulation of biologic products derived from bioengineered plants. Curr Op Biotechnol 2001;12(3):308-311.
33. Cramer C. Genetic Stability. Plant-Derived Biologics Meeting 2000 apr. (En línea) Disponible en: http://www.fda.gov/cber/minutes/plnt1040500.pdf.
34. Hammond J. Shed and Spread of transgenes. Plant-Derived Biologics Meeting 2000 apr. (En línea) Disponible en: http://www.fda.gov/cber/minutes/plnt1040500.pdf.
35. Cardineau G. Engineered Plants. Plant-Derived Biologics Meeting 2000 apr. (En línea) Disponible en: http://www.fda.gov/cber/minutes/plnt1040500.pdf.
36. Richter L. Potency, Consistency and Stability. Plant-Derived Biologics Meeting 2000 apr. (En línea) Disponible en: http://www.fda.gov/cber/minutes/plnt1040500.pdf.
37. Sasson A. Cultivos Transgénicos: hechos y desafíos. 1ra. edición. La Habana: Elfos Scientiae 2001).
38. CECMED. Regulación 40/2006: Requisitos químico-farmacéuticos y biológicos para el registro de productos biofarmacéuticos obtenidos a partir de plantas transgénicas, 2006 abr. (En línea) Disponible en: http://www.cecmed.sld.cu/Docs/RegFarm/DRA/LicProd/2003-2006/Reg/Reg_40-06.pdf.
39. Agencia de Información Nacional. Aprueban anticuerpo monoclonal obtenido a partir de plantas transgénicas. Periódico Granma (nota de prensa emitida por el CECMED),2006 jun 21. (En línea) Disponible en: http://www.granma.cubaweb.cu/2006/06/21/nacional/artic09.html.
40. Knäblein J, Pujol M, Borroto C. Plantibodies for human therapeutic use. BioWordl Europe 2007;1:14-7.
41. Sparrow PAC, Irwin JA, Dale PJ, Twyman RM, Ma JKC. Pharma-Planta: Road testing the developing regulatory guidelines for plant-made pharmaceuticals. Transgenic Res 2007;16:147-61.
42. SpöK A. Molecular farming on the rise-GMO regulators still working a tightrope. TRENDS Biotechnol 2007;25(2):74-82.
Received in October, 2008.
Accepted for publication in October, 2008.
Yanet Hechavarría Nuñez. Centro para el Control Estatal de la Calidad de los Medicamentos, CECMED Calle 200 No. 1706 e/ 17 y 19, Reparto Siboney, CP 11600, AP 16065, Ciudad de La Habana, Cuba. E-mail: yanet@cecmed.sld.cu

{kind=link}