










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBiotecnología Aplicada
versión On-line ISSN 1027-2852
Biotecnol Apl v.27 n.4 La Habana oct.-dic. 2010
REVIEW
Cellular and molecular insights into the wound healing mechanism in diabetes
Particularidades celulares y moleculares del mecanismo de cicatrización en la diabetes
Jorge Berlanga-Acosta1, Calixto Valdez-Pérezs2, William Savigne-Gutierrez2, Yssel Mendoza-Marí1, Neobalis Franco-Perez2, Evaristo Vargas-Machiran2, Natalia Poll-Marrón2, Hector Alvarez-Duarte2, Héctor Echeverria-Requeijo3, Rosa M Perez-Aguilar4
1 Center for Genetic Engineering and Biotechnology. CIGB Ave. 31 / 158 and 186, Playa, PO Box 6162, Havana, Cuba
2 Instituto Nacional de Angiología y Cirugía Vascular Calzada del Cerro # 1551, Cerro, Havana, Cuba
3 Hospital Clínico Quirúrgico Hermanos Ameijeiras, Havana, Cuba
4 Hospital Juan Manuel Márquez
ABSTRACT
Impaired healing in diabetes affects the resolution of both acute and chronic wounds. The vicious circle between wound chronicity and a deficient control of local infection is the cause that diabetic patients constitute 85% of all non-traumatic lower extremity amputations. From an etiological viewpoint, hyperglycemia is what triggers the onset and progression of biochemical disturbances that lead to systemic complications. In contrast to normal wound healing, physiological apoptotic clearance of inflammatory cells is prevented and the inflammatory phase is abnormally prolonged in diabetic wounds. Pro-inflammatory cytokines as tumor necrosis factor-alpha (TNF-a) and interleukin-1b (IL-1b) are increased in diabetic wounds with negative local and remote consequences. The etiopathogenic network consisting of inflammatory cytokines, local proteases, reactive oxygen and nitrogen species produces a cytotoxic and pro-degradation environment within the wound bed that impairs granulation and re-epithelialization. The nonenzymatic glycation of proteins, generating advanced glycation end-products (AGE), acts as an active pathogenic stream affecting healing. The accumulation of AGE interferes with DNA replication, cell anchoring, migration and proliferation. The binding of AGE to a receptor model (RAGE) may completely hamper the healing process. Diabetes impairs the recruitment and differentiation of bone marrow-derived stem cells, thereby limiting the availability of tissue repair cells. Re-epithelialization is also hindered by incomplete activation and/or differentiation of keratinocytes that impair migration. Novel and revolutionary pharmacological interventions are urgently needed to reduce diabetes complications, such as amputations of the lower extremities.
Keywords: diabetes, ulcer, amputation, granulation, re-epithelialization.
RESUMEN
El deterioro de la cicatrización en la diabetes afecta la resolución tanto de heridas agudas como crónicas. Se establece un círculo vicioso interamplificativo entre el fenotipo crónico y el control deficiente de la infección, que determina que el 85% de todas las amputaciones no traumáticas de miembros inferiores se practiquen en individuos diabéticos. La hiperglicemia es el detonador etiopatogénico proximal en el inicio y progresión de los desórdenes bioquímicos que dan lugar a las complicaciones sistémicas. Contrario a lo que ocurre durante la cicatrización normal, la eliminación apoptótica fisiológica de las células inflamatorias se detiene, lo que provoca un anormal estancamiento de la fase inflamatoria en las heridas diabéticas. En estas, además existe una sobre-expresión de citocinas pro-inflamatorias como el factor de necrosis tumoral alfa (del inglés, TNF-a) y la interleucina-1b (IL-1b), lo que trae consigo consecuencias deletéreas de impacto local y remoto. La red etiopatogénica de citocinas inflamatorias, proteasas locales, especies reactivas al nitrógeno y al oxígeno, propician un ambiente citotóxico y pro-degradativo en el lecho de la herida, que perjudica la granulación y re-epitelización. La glicosilación no enzimática de proteínas persiste como un ingrediente patogénico activo en el deterioro del proceso de cicatrización. La acumulación anormal de productos glicosilados interfiere con la replicación del ADN, el anclaje, la migración y la proliferación celular. La diabetes afecta la liberación, el reclutamiento y la diferenciación de las células madre derivadas de médula ósea, lo que limita la disponibilidad de estas células para reparar el tejido. La re-epitelización también se altera debido a la activación y/o diferenciación incompleta de los queratinocitos, lo cual obstruye su migración. Se necesitan de forma urgente abordajes farmacológicos novedosos y revolucionarios para reducir las diversas complicaciones de la diabetes, tales como la temida amputación de miembros inferiores.
Palabras clave: diabetes, úlcera, amputación, granulación, re-epitelización.
INTRODUCTION
Amputations are often considered to be the beginning of the end for patients with diabetes, since lower extremity ulceration is one of the several serious longterm complications associated to DM. The deficient healing of soft peripheral tissues leads to the onset of lower extremity ulcerations (1-3).
The term "diabetic foot" defines the specific features of the feet of diabetic patients, differentiating it from other conditions affecting the lower extremities. Infection, ulceration and the destruction of deep tissues associated with neurological abnormalities and diverse degrees of peripheral vascular disease at the lower limbs define diabetic foot (4). Although both neuropathy and vasculopathy, as individual entities, may co-exist and interact in diabetic foot, substantial clinical and histological differences can be distinguished between neuropathic and ischemic ulcer beds. Thus, neuropathy and hypo-perfusion of lower extremity tissues are long-term complications of hyperglycemia, which together with wound size and the host´s inability to fight local infection, determine the prognosis and the outcome.
Throughout evolution, wound healing has been a mechanism favoring the urgent structural and functional restoration of an injured area; it occurs as an innate cellular response to injury by involving two major cell functions: (1) the response of tissue-promoting cells and (2) the transient infiltration and homing of inflammatory cells. Diabetic wounds are a therapeutic challenge since we are dealing with the gross clinical expression of an enormous array of biochemical disturbances that have progressively undermined elementary biological mechanisms.
Here we examine the current knowledge of the biology of diabetic wounds, particularly the pro-inflammatory arm and the toxicity produced by the burden of the accumulation of advanced glycation end-products (AGE), leading to the onset of the hard-to-heal phenotype. The data here shown was selected from 910 reviewed papers downloaded from Pubmed and Bioline International (www.bioline.org.br) data bases through a direct search or through the Reference Manager program. Articles were retrieved using the following key restriction criteria: (1) Diabetic ulcer + inflammatory infiltrate, (2) Diabetic ulcer + granulation tissue, (3) Diabetic ulcer + epithelialization, (4) Diabetic ulcer + AGE, (5) Diabetes + RAGE + complications, (6) Diabetic ulcer + angiogenesis, and (6) Diabetic ulcer + growth factors.
GLUCOTOXICITY
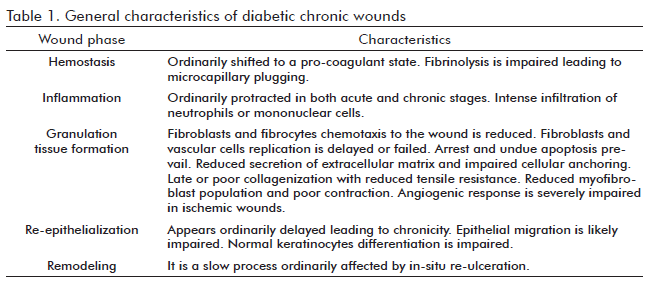
Although the onset and magnitude of diabetes complications are largely influenced by individual genetic factors, hyperglycemia is what triggers the cascade of complications (5). The difficulty in initiating and/or sustaining a physiological repair mechanism is one of the worst complications (6) since diabetes impairs most, if not all, of the events involved in the healing process (Table). The ulcer healing process is further complicated when lower limb hypoxia is added to the long-term hyperglycemia-derived toxicity (7). Chronic glucotoxicity affects most of the economy cells including those involved in the repair mechanism as shown by in vitro and in vivo studies under short-term or long-term exposure. Glucose toxicity is associated but not limited to: an increasing level of superoxide anions, impaired nitric oxide (NO) synthesis and subsequent depletion, the inhibition of protective and self-defense mechanisms of cells, the induction of DNA damage and distribution abnormalities (8). A general expression of this toxic effect is the resistance of cells to divide. The high level of glucose stops the production of endothelial and fibroblastoid cells; although the pathways leading to cell cycle arrest may differ between lineages (9), a common toxic effector appears to be the generation of reactive oxygen species (10). Glucose overload has been proven to inhibit endothelial nitric oxide synthase activity by mitochondrial superoxide production, it also imposes a pro-inflammatory program which may amplify insulin resistance and favor the onset of a chronic inflammatory response phenotype (11;12). As later shown, the systemic pro-inflammatory environment favors insulin resistance and often leads to more toxicity through hyperinsulinemia, forming a toxic vicious circle. The glucotoxicity-derived loops of oxidative stress, pro-inflammation, and the accumulation of terminal glycation products (commonly known as AGE - from advanced glycation end-products), disrupts the local homeostasis and depletes the wound cells of anti-oxidant defense resources and of growth factors and their receptors.
{kind=link}
THE INFLAMMATORY ENVIRONMENT IN THE DIABETIC WOUND
A chronic diabetic wound must be considered a proinflammatory organ placed in a metabolically deregulated host. The pool of wound-derived cytokines is enriched in the central circulation and blocks the action of insulin by phosphorylating key substrate proteins (13).
Tissue injury causes the immediate onset of acute inflammation. The inflammatory response is characterized by patterns of several leukocyte subsets that change in space and time and the well-defined chronology of the response is essential for optimal repair (14). The diabetic wound does not show the orderly cascade of events that characterizes normal wound healing; in contrast, the inflammatory reaction in diabetic wounds is prolonged. Serial biopsies from both neuropathic and ischemic ulcers-derived granulation tissue have indicated important histological differences, and therefore interpretative differences, between them in the absence of infection. The infiltration of neutrophils is intense, prolonged and not topographically polarized particularly in neuropathic wounds. It is not uncommon to observe a chronic infiltration preceded by an "acute" inflammatory cell, co-existing with a poor accumulation of extra-cellular matrix (ECM) in which collagen deposit is poor. In contrast, a widespread infiltration of round cells prevail in patients suffering from bed wound ischemia. These observations lead to the consideration that the biochemical microenvironment in ischemic and neuropathic ulcers is different and that the inflammatory "badge" corresponds to the most prevalent pathogenic component of the wound.
Although chronic diabetic ulcer is installed during the inflammatory phase of the normal healing process, this inflammatory reaction does not necessarily imply a local physiological control of bacteria. Experimental studies have shown that macrophages from diabetic mice phagocyte the cell detritus at a much slower rate and much less efficiently than their non-diabetic counterparts (15). Diabetic individuals are more susceptible to both wound infection and hyper-inflammation which can not be pathogenically separated from the elevated levels of pro-inflammatory cytokines as tumor necrosis factor a; (TNF-a) and interleukin 6 (IL-6) (16). Immune-related diabetes seems to occur because high glucose concentrations substantially disturb celldependent responses, whereas the correction of hyperglycemia improves leukocyte chemotaxis (17).
Pro-inflammatory cytokines are strongly up-regulated during the inflammatory phase. Data derived from diabetic rodents have shown a deregulated expression of macrophage inflammatory protein-2 (MIP-2) and macrophage chemoattractant protein-1 (MCP-1) which is associated with the increased and protracted infiltration of both neutrophils and macrophages into the wound (18). Compelling evidence indicates that neutrophils are critical for the acquisition of a prodegradative phenotype resulting from the imbalance between matrix synthesis and the degradation by stimulating the synthesis of matrix metalloproteinases (MMPs) (19). In line with this, TNF-a and interleukin 1b (IL-1b) secreted by neutrophils trigger signals for MMPs expression via the nuclear factor kappa B (NFkB) common pathway. Within the context of the wound, molecular targets of MMPs are numerous, and include not only elements of the ECM, but also locally secreted growth factors and their receptors. The observation that diabetic wounds are enriched in MMPs support the assumption that impaired growth factor availability may limit healing (18, 20, 21). Prolonged neutrophils infiltration is also linked to the overproduction within the wound area of elastase, reactive oxygen species (ROS) and reactive nitrogen species (RNS); all with a remarkable cytotoxic and pro-degradative potential (22, 23). In fact, high circulating and neutrophil-associated elastase levels are attributable to a poor glycemic control and are currently considered as risk markers for the development of diabetic angiopathy (24). Fibronectin degradation for instance, is referred as one of the several causes of diabetic reepithelialization failure because epidermal keratinocytes require the interaction between fibronectin and its surface receptor integrin a5b1 to effectively migrate (25). Curiously, insulin-degrading activity has also been demonstrated in the fluid of diabetic wounds which have been shown to correlate with the glycated hemoglobin levels. This evidence again highlights the importance of a strong metabolic control to ensure a linear healing process (26).
In contrast to the increased MMP-8 and 9 displayed by the non-healing diabetic wound, the concentration of NO is significantly reduced. Diabetic skin fibroblasts treated with NO donor compounds increased cell proliferation, and decreased the expression of MMP-8 and 9 in a dose-dependent manner. Thus, the fact that NO resumes the cell proliferation program and promotes the reestablishment of an anti-proteases effect is an argument in favor of beneficial effect of NO in wound healing (20).
An explanation for the abnormal diabetic pro-inflammatory reaction suggests that inflammatory cells evade apoptosis and thus extend the wound bed homing in a non-physiological manner. Although certain forces seem to prevent apoptosis of the inflammatory cells, other cells that are essential in granulation tissue growth are extremely prone to committing suicide (27). TNF-a has been largely involved in this controversial event. In addition to MMPs, high levels of TNF-a in the wound have been identified as a predictive molecular factor for wound closure failure (18). Type 2 diabetes is associated with high serum levels of inflammatory cytokines such as TNF-a (28). Within the wound context, TNF-a stimulates its own secretion and that of IL-1b, which contributes to a persistent inflammatory status (29) pushing the wound toward a catabolic slope (30). In line with this, TNF-a application causes a decrease in the tensile strength of the wound by reducing the expression of collagen types I and III (31;32). In general, there is a sharp antagonism between the pro-synthetic role of transforming growth factor b1 (TGF-b1) and the opposite TNF-a effect in terms of ECM deposition, wound contraction and maturation. The latter, through the c-Jun N-terminal kinase (JNK) pathway, inhibits Smad phosphorylation, reducing the transcription of TGF-b1, collagen 1A, fibronectin, and alpha-smooth muscle actin (a-SMA) (33). In fact, activation of the JNK pathway reduces insulin gene expression and interferes with insulin action. The suppression of this pathway in obese diabetic mice can protect cells from oxidative stress, and could therefore be a potential therapeutic target for diabetes. Conversely, the genetic ablation of the TNF-a receptor-1 globally improves the wound healing response by enhancing angiogenesis, collagen production, and re-epithelialization (34). The systemic administration of neutralizing antibodies against TNF-a into wounded ob/ob mice triggered a complete re-epithelialization (35).
TNF-a deregulation appears to contribute to the amplification of the apoptotic process observed in some diabetes-associated complications (34). Recent studies have illustrated the pro-apoptogenic effect of wound-secreted TNF-a on fibroblast populations in experimental diabetic wounds, while the intervention with a TNF-a specific inhibitor reduced caspase 3 activity and fibroblast apoptosis by almost 50% and the capacity for the formation of new matrix by 72% (34). The link between diabetes and apoptosis has been further strengthened by the identification of 71 over-expressed genes in diabetic animals that directly or indirectly regulate apoptosis and that significantly stimulate caspases activity. Converging evidence indicates that there is a remarkable up-regulation of apoptosis in granulation tissue-producing cells in wounds from individuals with poorly controlled blood sugar and concomitant microangiopathy (36). TNF-a has also been involved in the pathogenesis of micro and macrovascular pathology (37). Thus, the high level of fibroblastic cell apoptosis is a meaningful contributing factor for a deficient healing response in diabetic individuals. It is also likely that the TNF-mediated insulin resistance in the wounded tissue cells (38) could behave as a pro-apoptogenic factor. Accordingly, an anti-TNF-a neutralizing intervention restored insulin sensitivity and improved the healing process (39).
As shown by in silico simulation methods, any therapeutic approach aimed toward neutralizing TNF-a, or increasing active TGF-b1, would be similarly effective regardless of the initial assumption of the underlying disarrangement in the ulcerogenic process (40).
Chronic wounds, and especially diabetic foot ulcers, have a highly pro-oxidant microenvironment which complements and amplifies the pro-inflammatory arm in the cytotoxic cascade. Diabetic tissues produce abnormal amounts of ROS and are, at the same time, the victims of their attack. In a systemic context, ROS contribute to the onset of insulin resistance by inactivating the signaling pathway between the insulin receptor and the glucose transporter system (41). Leukocytes, especially neutrophils are a rich source of various reactive species which are released into the wound environment. Endothelial cells and fibroblasts, particularly senescent fibroblasts, are a prominent population in chronic wounds, but also a potential source of ROS. Thus, the disturbed oxidant/antioxidant balance within the chronic wound is considered a major factor in the amplification of the inflammatory state (42) in regard to the deficient availability of growth factors and functionality (43). Conclusively, TNF-a inhibition is apparently sufficient to neutralize the misbehaving inflammatory machinery in non-healing wounds, so as to assist in reprogramming the whole local microenvironment.
THE CYTOTOXIC EFFECT OF THE ADVANCED GLYCATION END-PRODUCTS (AGE)
Mounting evidence indicates that the biology of the AGE-RAGE system in diabetic individuals is a molecular trigger and amplifier of most, if not all, of the accumulative disease complications, including impaired healing (44). Within Brownlee´s "Unifying Hypothesis" AGE are in second place within the apparently distant pathogenic pieces. Hyperglycemia seems to be a major requirement in the non-enzymatic glycation process that generates the heterogeneous group of cytotoxic AGE compounds. Consequently, ROS are formed along with the AGE generation process, and correspondingly, ROS hasten AGE formation, thus paving the way for a self-perpetuating pathogenic cycle of ROS-AGE which appears to characterize the biochemistry of diabetes (45-47). The accumulation of AGE in cells exposed to chronic hyperglycemia and oxidative stress result in irreversible damages even when these cells return to a normal glycemic environment (45, 46, 48-52).
AGE acts through a cell surface receptor that coincidently appears to be up-regulated in most tissues of diabetic patients. Different receptors for AGE have been discovered, one of these, termed RAGE (Receptor for Advanced Glycation End-Products), initiates the intracellular signaling that disrupts cellular function. Nuclear factor kappa-B (NF-kB) binding sites, an interferon-g response element, and a nuclear factor-interleukin-6 DNA binding motif, are located on the RAGE promoter region. The fact that NF-kB controls the cellular expression of RAGE establishes a functional link between RAGE and the inflammatory response (53;54).
AGE-RAGE interaction triggers the generation of pro-inflammatory cytokines, adhesion molecules, and chemokines, thus enhancing the attraction of more inflammatory cells? and perpetuating the inflammatory profile (55). Up-regulation of RAGE has been described on endothelial, smooth muscle cells and mononuclear phagocytes; while AGE tends to accumulate in non-labile dermal cutaneous proteins as collagen and elastin, thus meaning that the skin of diabetics is physicochemically altered. Under the microscope, diabetic skin have degenerative changes observed as loosely arranged collagen and increased apoptotic cells. The immunohistochemical analysis of the granulation tissue of diabetic ulcer patients indicates the accumulation of AGE and RAGE co-expressed in productive endothelial cells with frustrated angiogenesis. Other coexisting microscopic changes are cytoplasm degeneration, the inability to line up and to round up as to establish an appropriate co-opting for a vascular collar. These chemical and physical changes occur in human skin collagen with age and appear to speed up with diabetes (56). AGE cross-linking reactions in collagen also contribute to diabetic circulatory complications such as dermal and vascular stiffening. This was confirmed by the immediate toxic consequences of AGE accumulation in rats challenged with methylglyoxal (MGO); a well-identified AGE precursor. The animals abundantly reproduced most of the diabetic systemic complications but within a normal glycemic environment throughout the experimental period. It was shown that the uncontrolled prevalence of AGE/RAGE impairs the appropriate balance required between antagonistic forces in physiological processes such as: pro-inflammation/counter-inflammation, regeneration/ degeneration, and vasodilation/vasoconstriction. The study also demonstrated that in the environment of intense AGE accumulation, the granulation tissue of the wound histologically reproduced features of the human diabetic wound bed. It was furthermore demonstrated that the granulation tissue of the MGO-treated animals had turned into a local pro-inflammatory organ due to the intense expression of immuno- labeled TNF-a and IL-1b (57). This concept has recently been extended to the periodontal tissue healing process. Again, MGO treatment efficiently glycated collagen and fibronectin, transformed the gingival fibroblast biology and impaired the healing process mirroring the periodontal healing failure observed in diabetic patients (58). This demonstrates the deleterious involvement of MGO-AGE-RAGE in diabetic tissue damage (59). Other experiments show that diet-derived AGE significantly affects the rate of wound healing, which is associated with AGE skin deposit (60). AGE interacts with dermal fibroblasts and endothelial cells (61;62), while both cells express RAGE and are sensitive to ligand interaction. Eventually they show apoptosis (63). Recent studies have established a signaling pathway evolving to fibroblast apoptosis through the activation of p38 and JNK pathways, leading to an enhanced caspase-3 activity and FOXO1 transcription factor (64). This and other evidence currently place the AGE-RAGE axis at the center of the toxic and proinflammatory cascade of events that disturbs wound healing in diabetes (65). In summary, AGE-RAGE interaction is so deleterious that it facilitates the onset of pro-apoptotogenic, pro-inflammatory, pro-oxidant and pro-degradative wound environment.
GRANULATION AND RE-EPITHELIALIZATION PROCESSES ARE IMPAIRED IN DIABETES
It is known that diabetes is accompanied by the delayed and/or insufficient production of granulation tissue. Findings show that diabetic ulcer fibroblasts are morphologically different from their healthy counterparts.
Diabetic fibroblasts are larger and widely spread. Under transmission electron microscopy these cells reveal a large dilated endoplasmic reticulum, a lack of microtubular structures and multiple lamellar and vesicular bodies. This abnormal morphology provides the theoretical basis for a decreased proliferative capacity and other abnormal traits (66, 67). It has not been established if these structural changes are secondary to a long-term exposure to hyperglycemia and the general cytotoxic milieu. But it has been historically well-documented that high glucose concentrations inhibit fibroblast proliferation and induces growth factor resistance, which tends to explain their proliferative failure. We have detected an enhanced expression of the anti-proliferative, pro-senescent protein (68), prohibitin in granulation tissue fibroblasts of diabetic foot ulcers as compared with healthy subjects repairing a second degree burn (Jorge Berlanga, unpublished). Thus, it is accepted that fibroblasts derived from chronic diabetic ulcers have lower intrinsic proliferative capability than those collected from intact skin areas (69). To further substantiate this assertion, it has been shown that cultured fibroblasts from diabetic patients require the presence of multiple supplements in addition to growth factors for their proliferation while other data suggest a deficit of certain growth factors receptors involved in cell proliferation (70). Dermal fibroblasts from diabetic mice exhibit abnormalities even when grown in an ex vivo culture environment that has been optimized for nutrients, growth factors, and glucose concentrations. Different in vitro assays, including hypoxic or normoxic conditions, show that the diabetic fibroblast population does not physiologically migrate as does its healthy counterpart, and thus becomes hyporesponsive to the hypoxic challenge (71, 72). These cells are also more prone to ischemiainduced apoptosis and to up-regulate p53 expression than the non-diabetic controls. They are also unable to up-regulate the Vascular Endothelial Growth Factor (VEGF) production under hypoxic conditions whereas wild-type fibroblasts show a several fold increase of VEGF under the same stressing conditions (73).
Under hypoxic conditions, Hypoxia Inducible Factor-1a (HIF-1a) is stabilized against degradation and up-regulates a series of genes involved in angiogenesis, glycolytic metabolism, cell proliferation, and survival. Studies have shown that HIF-1a protein levels are dramatically reduced in wounds from diabetic mice as compared to their non-diabetic littermates (74-76). Hyperglycemia impairs the hypoxia-dependent stabilization of HIF-1a against proteasomal degradation in primary human dermal fibroblasts, human microendothelial cells, as found in foot ulcer-derived cells (77). It has been determined that this HIF-1a reduction accounted for a decreased DNA-binding activity and a reduced expression of several downstream target genes, including VEGF. Conversely, the induction of a sustained HIF-1a expression significantly restored the wound healing process (78).
The fibrocyte, a bone marrow-derived mesenchymal progenitor cell appears to contribute to the healing process by enriching the population of fibroblasts and myofibroblasts (79). Laboratory evidence (80, 81) leads to the hypothesis that fibrocyte wound recruitment and differentiation is reduced in diabetes, acting as a limiting factor for successful and progressive granulation.
Re-epithelialization in mammals is far more complex and much slower than in lower organisms, and demands the combined action of multiple factors for keratinocyte migration and proliferation. Although reepithelialization failure has been largely recognized as an essential feature of diabetes and other chronic wounds, its molecular basis must still be fully elucidated. All pathologists distinguish the epidermis of a chronic wound edge as a thick and hyperproliferative structure with mitotically active keratinocytes that are apparently unable to migrate along the surface. It has therefore been speculated that the non-healing edge keratinocytes do not successfully complete either of two possible pathways: activation or differentiation (82). In consonance with this, one of the main issues in chronic wound treatment is how to revert the keratinocytes phenotype into a proper differentiating and migratory program (82).
The first scientific indication that insulin is biologically relevant for skin cells derives from the fact that insulin is an essential component for human keratinocyte culture, demonstrating its involvement in the regulation of proliferation, apoptosis, and metabolism (83, 84). Recent studies in this field show that insulin contributes to the release of VEGF in skin wound cells through an Akt1-mediated post-transcriptional mechanism (85). Glucose is known to affect insulin action by regulating the expression of several genes including the insulin receptor at both the transcriptional and translational levels (86). The lack of an insulin receptor expression results in reduced skin proliferation and abnormal differentiation in vivo (87). Furthermore, glucose has been shown to have a direct toxic effect on keratinocytes. As for other cells grown in the presence of high glucose concentrations, human epidermal keratinocytes significantly reduce proliferation rate and replicating life span (88) and were found to be more susceptible to apoptosis (89). Other studies also demonstrated that hyperglycemic conditions abort the proliferative ability of keratinocytes and their migratory response (90). Aside from the glucose-mediated direct cytotoxic effect on the keratinocytes, the modification through AGE of type-I collagen and other ECM proteins impairs the integrin-mediated adhesion of keratinocytes to the basement matrix, and could thus contribute to the pa- thogenesis of diabetic re-epithelialization failure (91). In this context, epithelial-mesenchymal interaction plays a prime role in establishing the profile and order of released factors regulating proliferation and differentiation of keratinocytes (92).
Recent experiments have introduced another line of evidence that favors the roles of c-myc and b-catenin in impairing the migration of epithelial edges, involving mechanisms that may ultimately deplete the pool of epidermal stem cells at the non-healing edge (91). The routine practice of the sharp debridement of ulcers is a useful procedure for epithelial cells to resume their activation cycle and differentiation program by transforming chronicity to a more acute phenotype (93).
Classic experiments provide illustrative examples of the relevance of the epithelial-mesenchymal actions and on the irreplaceable role of growth factor as a networking bridge (94) in re-epithelialization. Skin-reconstitution studies have shown that bone marrow stromal cells (BMSCs), in addition to dermislocalized preadipocytes and fibroblasts, distinctively promote epidermal regeneration (95). As diabetes proceeds with a deficient secretion of growth factors and other chemotactic mediators in areas of tissue repair, the recruitment of circulating stromal cells may be reduced, which is an additional blow to the already existing high glucose-associated toxicity (96). Finally, TNF-a has also been involved in epithelial cell arrest by deeply perturbing critical elements of keratinocyte physiology (97).
CONCLUDING REMARKS AND OUTLOOK
Diabetes complications have a Malthusian behavior because of their unrivaled proportions in terms of morbidity and mortality. Even with optimal management, many individuals with diabetes become blind, develop renal failure and require amputation. The latter is the expression of two negatively cooperating forces: (1) a deteriorated mechanism for tissue repair and (2) an insufficient control of infection. It was surprising to see that high glucose concentrations are toxic per se to target cells such as cutaneous fibroblasts, even after an acute short-term exposure. Far less unexpected was the finding that insulin is a key hormone for skin cell physiology and homeostasis. Chronic hyperglycemia establishes a complex biochemical interconnection with some of its parallel pathophysiological derivatives, thus enriching the toxic profile of the diabetic individual´s environment. This context has an obvious deleterious impact in a process that requires a twofold or three-fold cell replication to recreate novel structures with a differentiated phenotype. There is a need to overcome major hurdles: (1) the ulcer is the consequence of the long-term damage of diabetes to skin tissue, nerves and vessels. (2) The architectural cells of granulation tissue are reluctant to proliferate and secrete, and become largely susceptible to apoptosis. Thus, the chronic phenotype is basically characterized by hypocellularity, deficient neomatrix synthesis- organization, cell cycle arrest and the onset of a senescent phenotype. Diabetes stamps some kind of metabolic memory as wound fibroblasts appear reluctant to proliferate even at optimal culture conditions. Understanding the mechanistic bases of this behavior could offer clues on the fundamentals of wound chronicity. Surgical debridement has always proved to be clinically useful. The former "chronic into the now acute" sHIFt is the consequence of genes moved upon debridement that restore cells´ proliferative advantages. Moreover, a major clinical challenge is how to keep these wounds within a synchronic linear healing trajectory. It is likely that the administration of exogenous growth factors could become a fueling force. As mentioned, diabetic ulcers show a huge failure in the physiology of the growth factors-receptors axis. The administration of natural or recombinant growth factors (single or combined) may be interpreted as a replacement therapy. It is likely that the therapeutic usefulness of growth factors stems from their ability to counter-balance a variety of cell-cycle inhibiting proteins that prevail in chronic wound G1-arrested cells.
The introduction of live skin equivalents have also shown clinical efficacy in the treatment of low-grade, neuropathic diabetic chronic wounds. They were found to significantly shorten healing time. These local bioreactors appear to nourish the wound cells with growth factors and ECM proteins that somehow ameliorate or prevent the deficient replication and proliferation that characterizes the diabetic wound milieu.
An important therapeutic area focuses on the down-regulation of the inflammatory process and its collateral effects. Thus far, the target has been TNF-a inhibition. The first evidence was provided by Doxycycline, an antibiotic that inhibits metalloproteinases so as to TNF-a converting enzyme (TACE). Doxycycline improved the healing of chronic diabetic foot ulcers. The data was supported when using the therapeutic anti-TNF-a neutralizing antibody (infliximab) that was topically administered and improved the healing of a series of chronic ulcers of multi-factorial etiology. Irrespective of the differences existing in these studies, TNF-a neutralization is the common biological concept.
Novel and even more revolutionary therapeutic concepts are expected. Of particularly significance, and not only for wound healing, is the pharmacological manipulation of the AGE-RAGE axis. An intervention leading to the reduction of circulating and in-tissue accumulated AGE may even reach prophylactic value. On the same strategic line is the use of a decoy factor to prevent RAGE activation. The hypothesis that a soluble RAGE would restore healing progression was experimentally validated years ago. The manipulation of the AGE-RAGE axis may target more that one etiopathogenic damage component. The integrality and polyvalence of such an intervention would certainly contribute to improve healing in diabetic patients.
REFERENCES
1. Most RS, Sinnock P. The epidemiology of lower extremity amputations in diabetic individuals. Diabetes Care 1983;6(1): 87-91.
2. Bild DE, Selby JV, Sinnock P, Browner WS, Braveman P, Showstack JA. Lowerextremity amputation in people with diabetes. Epidemiology and prevention. Diabetes Care 1989;12(1):24-31.
3. Sen CK, Gordillo GM, Roy S, Kirsner R, Lambert L, Hunt TK,