










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBiotecnología Aplicada
versión On-line ISSN 1027-2852
Biotecnol Apl v.28 n.1 La Habana ene.-mar. 2011
RESEARCH
The Sticholysin I mutants St I E2C and St I R52C show similar binding to liposomal vesicles but differ in their permeabilizing activity
Mutantes St I E2C y St I R52C con similar actividad de unión en vesículas liposomales y diferencias en la permeabilización
Aracelys López1, Aisel Valle1, Lohans Pedrera1, Diana Martínez1, Rafael Fando2, Shirley Schreier3, Carlos Álvarez1, María E Lanio1, Fabiola Pazos1
1 Center for Protein Studies, Havana University Street 25 #455 / J and I, Vedado, PO Box 10 400, Havana, Cuba
2 National Center for Scientific Research, CNIC Ave. 25 and 158 #15202, PO Box 12100, Havana, Cuba
3 Chemistry Institute, São Paulo University, Brazil
ABSTRACT
The mechanism of pore formation by actinoporins is a multistep process, involving binding of water soluble monomer to membrane and subsequent oligomerization of monomers on the membrane surface, forming a functional pore. However, molecular details of membrane insertion mechanism and oligomerization are not clear. A phosphocholine-binding site and a surface cluster of aromatic rings, together with a basic region, are important to the initial interaction with membrane and the N-terminal region is relevant in the pore formation. Aiming to deepen into the structure-function relationship in sticholysins, we designed and produced two Cys mutants of recombinant sticholysin I (rSt I) in relevant functional regions for membrane interaction: St I E2C (in the N-terminal region) and St I R52C (in the membrane binding site). Conformational studies suggested that the replacement of Glu-2 and Arg-52 by a Cys residue in rSt I not noticeably changes protein conformation as assessed by fluorescence and CD spectroscopy, the first change not affecting toxin’s permeabilizing ability. The relative decrease in the pore forming capacity of St I R52C is not related with a smaller binding capacity of this mutant to membrane. In summary, St I E2C and St I R52C retain the main conformational properties of the wild type and show similar binding to liposomal vesicles while differing in their permeabilizing activity. St I E2C and St I R52C constitute good tools to study those steps of the permeabilizing mechanism of sticholysins that take place after binding to membrane, using thiol-specific probes such as fluorescent and spin labels.
Keywords: pore-forming toxin, actinoporin, membrane-protein interaction.
RESUMEN
El mecanismo de formación de poros de las actinoporinas es un proceso de varias etapas hasta la formación de un poro funcional. Se requiere un sitio de unión a fosfocolina, un grupo de anillos aromáticos y una región básica, para la interacción inicial con la membrana. La región N-terminal es relevante para la formación del poro. En este trabajo se diseñó y obtuvo dos mutantes de Sticolisina I (St I) con Cys en regiones funcionalmente relevantes para la interacción con membranas: St I E2C (en la región N-terminal) y St I R52C (en el sitio de unión a membranas). El reemplazo de los residuos Glu-2 y Arg-52 por Cys no produce cambios notables en la conformación de St I, según determinaciones de fluorescencia y espectroscopía de dicroísmo circular. El primer cambio no afectó la actividad permeabilizante. La disminución relativa en la capacidad formadora de poros en St I R52C no se vincula a su menor capacidad de asociación con la membrana. St I E2C y St I R52C conservan las principales características conformacionales de St I nativa y muestran similar capacidad de unión a vesículas liposomales, mientras que difieren en cuanto a actividad permeabilizante. Estos dos mutantes son herramientas útiles para estudiar los pasos de los mecanismos de permeabilización de las Sticholisinas, en especial los que ocurren tras su unión a las membranas, mediante el uso de sondas específicas para grupos tiol tales como indicadores fluorescentes y del momento del espín.
Palabras clave: toxina formadora de poro, actinoporina, interacción membrana-proteína.
INTRODUCTION
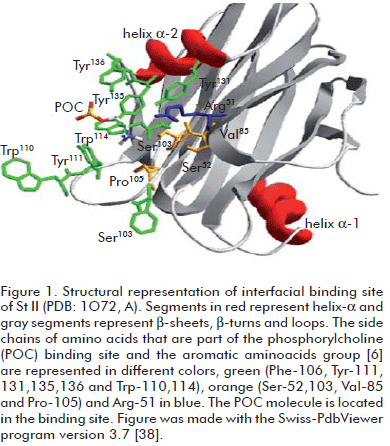
Sticholysin I (St I) and Sticholysin II (St II) are two isotoxins obtained from the sea anemone Stichodactyla helianthus (1). Sticholysins belongs to the actinoporin family, which is constituted by pore-forming proteins isolated from sea anemones. St I is a cysteineless and basic protein of 20 kDa that increases ion membrane permeability by forming a channel resulting from the association of three or four toxin monomers (2). The three-dimensional structures of three actinoporins, equinatoxin II (Eqt II) from Actinia equina (3, 4) and St I (5) and St II (6) are now known. These toxins display a very similar β-sandwich fold flanked on each side by two short α-helices. The structure of a complex St II: phosphocholine (POC) allowed postulating a POC binding site for the actinoporin family (6). The POC binding site is formed by side chains of Ser-52, Val-85, Ser-103, Pro-105, Tyr-111, Tyr-131, Tyr-135 and Tyr-136 (Figure 1). These residues are almost completely conserved within the actinoporin family, indicating that POC binding may proceed in the same way in all actinoporins (7).
Actinoporins are able to spontaneously insert themselves into model and natural membranes and form oligomeric pores. The mechanism for pore formation involves several steps. First, the monomer toxin is attached to the membrane by the specific recognition of sphingomyelin (SM) using the aromatic rich region and adjacent POC binding site (1, 3, 8, 9). Then, the N-terminal segment is transferred to the lipid-water interface (8, 10, 11) and finally the toxin oligomerizes on the membrane surface and α-helices of 3 or 4 monomers insert into the bilayer forming the functional pore (2, 11-13). In the last decade, research on actinoporins has rapidly expanded, thereby providing more profound insights into their structure, molecular mechanisms and their probable biotechnology or biomedical applications. For instance, they were used for the selective killing of parasites (14) and cancer cells (15-18), with built-in biological ‘triggers’ that would activate in response to specific biological stimuli, or as biosensors (19, 20).
Thus, in order to gain new insights into the role of certain sticholysins residues during the pore formation process, we designed and produced two Cys mutants in relevant functional regions of recombinant sticholysin I (rSt I): St I E2C (in the N-terminal sequence) and St I R52C (near the membrane binding site) (8, 21, 22). The overall results suggest that the replacement of Glu-2 and Arg-52 by a Cys residue in rSt I did not noticeably change protein conformation at least from the data derived from fluorescence and CD spectroscopic studies. Furthermore, the substitution of an amino acid by Cys, either close to the membrane binding region (St I R52C) or in the N-terminal region (St I E2C), did not change their ability to bind to liposomal vesicles, while producing a decrease in St I R52C pore-forming activity. These results clarify that Arg-52 is relatively more important for pore formation than binding in St I.
MATERIALS AND METHODS
Mutagenesis, expression and purification of the mutants
Positions for mutation were selected considering their location in the relevant protein regions for sticholysin function (22) and exposure to the solvent of each residue in the protein structure. Accessible surface area (ASA) of residues in the rSt I three dimensional model (23) was calculated as ASA of amino acid (X) in the rSt I structural model relative to a Gly-X-Gly tri-peptide in a vacuum employing the WhatIf server (http://swift.cmbi.kun.nl/whatif/).
Mutations were introduced by PCR using as a template vector pET3a-rSt I (23) and adequate oligonucleotides (Table 1). PCR was performed as previously described (23). The products of gene amplification were restricted with NdeI and BamHI and cloned into pET3a, where the nucleotide sequence of putative positive clones was verified using a T7 Sequencing TM Kit according to Pazos et al. (23). Mutants and wild type proteins were expressed in Escherichia coli BL21 (DE3) pLysS strain and purified from supernatants of lysed bacteria using ion-exchange chromatography on carboxymethyl cellulose (CM-52) (23). For mutants, an intermediate washing step with 100 mM β-mercaptoethanol was included before the gradient to eliminate molecules linked to the toxin through Cys (23). The homogeneity of proteins was verified by sodium dodecylsulphate-polyacrylamide gel electrophoresis (SDS-PAGE) (24) and reverse phase-high pressure liquid chromatography (RP-HPLC) on a Vydac reversed phase column RP-C4 (23). Pro- tein concentration was determined measuring absorbance at 280 nm using the extinction coefficient of St I (1).
Hemolytic activity assay
Hemolytic activity of toxins was evaluated from the decrease in turbidity of a human red blood cell suspension as previously described (25). The percentage of hemolysis was estimated from the final extension of the hemolytic process until 30 minutes after the start of the assay as follows:
![]()
where: ODi and ODf are the optical density values at the beginning and 30 minutes after the start of the hemolytic assay, respectively; and ODs represent the OD value corresponding to the total lysis of the erythrocyte suspension provoked by an excess of toxins.
Sphingomyelin (SM)
Large unilamellar vesicles (LUVs) were prepared by extruding multilamellar liposomes of palmitoyloleyl-phosphatidylcholine (POPC) and SM from bovine brain (Avanti Polar Lipids, USA) in a molar ratio of 85:15, prepared in the presence of 80 mM carboxyfluorescein (pH 7.0) according to the procedure described by Tejuca et al. (2). Extrusion was carried out with a two-syringe extruder (Lipo Fast Basic Unit, Canada), equipped with 100 nm polycarbonate filters (Nucleopore, USA). The non-encapsulated fluorescent probe was removed from the vesicle suspension by a Sephadex G-50 gel filtration column under isosmotic conditions. Small unilamellar vesicles (SUVs) were prepared by sonication of multilamellar vesicles comprised of POPC:SM (85:15) as described elsewhere (23). The final lipid concentration of the liposome suspension was determined according to Rouser et al. (26).
Intrinsic fluorescence measurements
Fluorescence measurements were recorded in a spectrofluorimeter (Shimadzu RF-540, Japan) using 1 cm path length quartz cuvettes. Slit widths of a nominal band pass of 5 nm were used in both excitation and emission beams. Intrinsic fluorescence emission spectra of 1.5 μM St I mutants and rSt I solutions were recorded from 300 to 450 nm after excitation at 295 nm to obtain fluorescence spectra only derived from tryptophan residues (27). Background intensities measured in samples without protein were always subtracted. Changes were also measured in the intrinsic fluorescence of St I mutants and rSt I upon the addition of increasing amounts of SUVs.
Permeabilization assays
Permeabilization was studied by measuring the fluorescence of carboxyfluorescein released from LUVs after adding different concentrations of proteins according to Tejuca et al. (2). The total fraction of permeabilized vesicles was determined as follows:
![]()
where: F0 and Ff represent the fluorescence values before and 10 minutes after the start of the assay and Fmax is the fluorescence value achieved after the addition of 1 mM Triton X-100.
Circular dichroism spectra
Far-UV and near-UV circular dichroism (CD) spectra of the St I mutants and rSt I were recorded on a CD6 Jobin Yvon spectropolarimeter (Longjumeau, France) coupled to a Multiscan Computer (D&D Technology) and base-line was corrected by control samples of similarly prepared solutions devoid of protein. Spectra were obtained for far-UV from 190 to 260 nm, and for near-UV range from 250 to 350 nm, in 1-mm and 5-mm path length quartz cuvettes, respectively. The reported spectra are averages from 6 and 12 scans for far-UV and near-UV, respectively.
RESULTS AND DISCUSSION
For a better insight in the pore formation mechanism and the structure-function relationship of sticholysins, we have designed and produced two Cys mutants of rSt I, taking advantage of the fact that these toxins are cysteine-less proteins (28). Glu-2 and Arg-52 were selected to be replaced by Cys because of the location in the N-terminal sequence and for being near the phosphocholine-binding site, respectively. Secondly, these residues have a relatively high exposure to solvent (85.5% for Glu-2 ASA and 50.7% for Arg-52 ASA) (29). The introduction of this reactive residue in the St I amino acid sequence would enable the modification of these mutants with thiol-specific probes and to study the location of the modified region in the protein-membrane interaction. Such an approach is valid only if these mutants retain the wild-type conformation and the ability to form a functional pore.
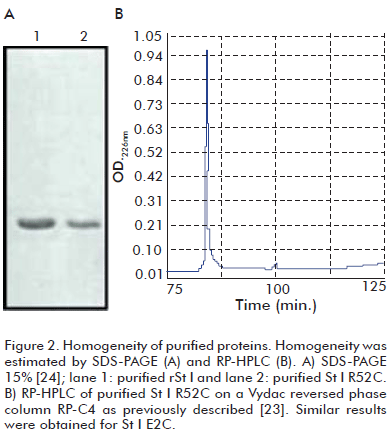
Putative positive clones selected by restriction analysis were sequenced to confirm the presence of the desired Cys mutations in the constructions and the integrity of the rest of the genes. The mutants were purified by a single step of ion-exchange chromatography. The homogeneity of toxins was higher than 95%, estimated by SDS-PAGE (Figure 2A) and RP-HPLC (Figure 2B). Protein concentration was about 10 μM in all experiments. The mutants were in a monomeric form, as demonstrated by SDS-PAGE (Figure 2A).
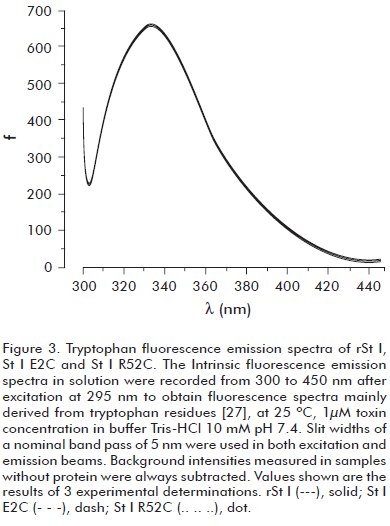
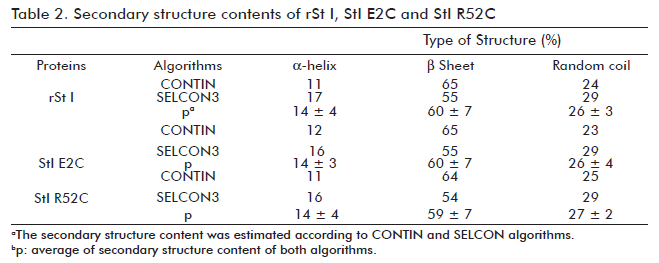
Before evaluating the functional activity of St I E2C and St I R52C, we verified that they conserved the wild type conformational properties. We used fluorescence and CD spectroscopy to rule out any significant conformational changes. The intrinsic fluorescence spectra of mutants in solution are very similar to the wild type (Figure 3). The far-UV CD spectra of the wild type and mutants are very much alike (Figure 4A), showing a positive band centered at 195 nm and a minimum around 217 nm, which is typical of proteins containing basically β-sheet structures (30). The relative secondary structure content of the toxins was established from their far-UV CD spectra using CONTIN and SELCON algorithms (Table 2). Hence, the mutations of selected residues did not modify St I secondary structure.
Also St I E2C, St I R52C and rSt I showed similar near-UV spectra, with the same main band positions, in spite of the presence of Cys in the mutant sequences, indicating that the three dimensional conformation of rSt I was conserved after mutation (Figure 4B). We observed only a slight difference between the spectra of rSt I and St I R52C. The spectrum of this mutant showed more pronounced negative bands at 266 and 270 nm, which are in the Phe range (31). These differences could be determined by the nearness of Phe-51 to the mutated position.
The pore formation ability of the mutants was measured by hemolytic activity experiments, which is the most widely used assay for testing the functional activity of actinoporins. rSt I and St I E2C showed similar hemolytic capacity (Figure 5). The molecular mechanism of actinoporin pore formation has been unraveled in the last few years with particular emphasis on the role of the N-terminal region. It was shown that it needs to be flexible (8, 32) and the region 14-23 in St II forms an amphiphatic α-helix (3), the only recognized structural element of the final pore thus far (11). Actinoporins with a removed N-terminus do not lyse red blood cells (33). Moreover, various tags added to the N-terminal part of these proteins decrease toxin activity (34, 35). Considering this, it could be possible to assume that a mutation in the second position in St I could modify its pore-forming ability. Nevertheless, this result indicates that the change of Glu-2 to Cys did not affect rSt I pore formation capacity in terms of hemolytic activity.
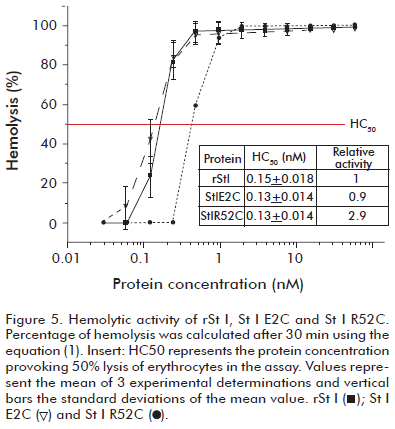
In contrast, the HC50 value of St I R52C is about three times higher than the HC50 value of rSt I, meaning a lower lytic capacity (Figure 5). This result suggests an important role of the Arg-52 residue in the functional capacity of this toxin. To further investigate the causes the lower functional activity of St I R52C and for an insight into St I-membrane interaction events, we used model bilayer systems to study the functional properties of the mutants.
We studied the binding ability of rSt I and its mutants using SUVs of POPC:SM (85:15). The binding step of toxins to lipid bilayers was assessed by measuring the change of protein intrinsic fluorescence upon the addition of increasing concentrations of SUVs. The addition of liposomes progressively increases fluorescence intensity until reaching a plateau indicating a quantitative association of proteins to lipid bilayers, where rSt I, St I E2C and St I R52C display a similar binding ability to SUVs (Figure 6). Adjusting the experimental data to the Boltzman function we obtained the value of parameters Lip50 (amount of lipid needed to bind half of the protein present in the assay) and Fmax/F0 (ratio of fluorescence values in the presence of infinite lipid concentrations and in the absence of lipids). As expected, the values of these parameters for the toxins are very similar, indicating similar membrane binding ability (Figure 6). This result shows that mutations did not affect the binding capacity of St I under these experimental conditions.
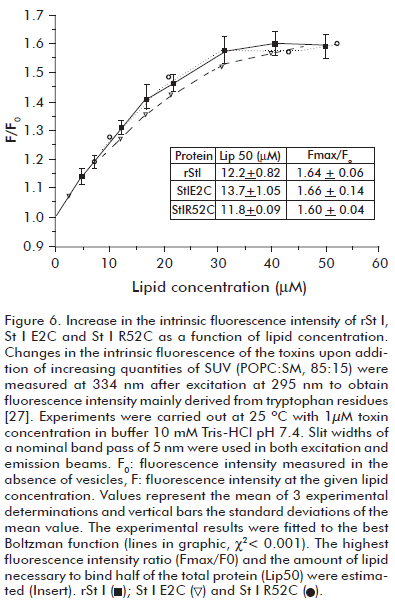
It is interesting to note that Arg-52 is located near the phosphorylcholine binding pocket (6) (Figure 1) and a change in this position could modify the final lodging of the toxin in the lipid bilayer. However, the experimental assay used was not able to detect the differences in the membrane binding ability between rSt I and its mutants. Recently, Castrillo et al. (36) studied the binding of St I to dodecylphosphocholine micelles by NMR. They did not report interaction between the micelles and Glu-2 or Arg-52 residues in the protein.
Pore formation by rSt I and its mutants was evaluated by their ability to release carboxyfluorescein encapsulated in LUVs. They promoted the exit of fluorophore entrapped in the inner pool of LUVs composed of POPC:SM (85:15). The molar ratio of the total fraction of permeabilized vesicles (f) vs. protein-lipid is illustrated in figure 7. The results of permeabilization assays show a good correlation with those observed in hemolytic activity (Figure 5). We obtained very similar curves for rSt I and St I E2C. In contrast, St I R52C showed relatively smaller permeabilization values than rSt I.

Arg-52 appears to be highly preserved in the sequence alignment of actinoporins (22) suggesting an important role of this residue in toxin function. Mancheño et al. (6) described the phosphorylcho-line group binding site in the St II three-dimensional structure as a partly hydrophilic and hydrophobic cavity due to the hydrophilic groups of Tyr and Ser residues, the aromatic ring of Tyr residues and the side chains of Val and Pro residues, located in this region (Figure 1). They proposed the role of the Arg residue arguing that its positive charge presumably contributes to the stabilization of the binding of the phosphocholine group with the protein. Therefore, the substitution of this residue by a Cys could change the properties of the phosphorylcholine binding si- te, modifying the interaction of the toxin with the membrane.
Nevertheless, the relative decrease in pore forming capacity of St I R52C (Figures 5 and 7) is not related to a lower binding capacity of this mutant to the membrane (Figure 6). Hence, this residue could be involved in steps occurring after St I binding in the pore formation events. Similar results were reported by Anderluh et al. (37) during the functional characterization of the Eqt II S54C mutant. This mutant showed lower hemolytic and permeabilizing activity than the wild type, but displayed a capacity to bind LUVs that was similar to Eqt II. Ser-54 in Eqt II is the equivalent residue to Ser-53 in St I, which is the Arg-52 adjacent amino acid. Considering the spatial nearness of these two residues and the similar behavior of their respective mutants, the participation of the Arg-52 residue in a step different to that of the toxin bilayer binding could be strongly considered although this requires further experimental studies.
On the other hand, although the mutation at position two of the St I amino acid sequence is located in the N-terminal region, and probably involved in the transmembrane pore formation, this mutation did not affect the pore forming ability of the toxin. Kristan et al. (21) demonstrated that the most N-terminal portion of Eqt II extends to the membrane trans side in the final pore conformation. Moreover the Eqt II S1C mutant was able to retain the pore forming capacity of Eqt II (37). An examination of the amino acid sequence alignment of the actinoporin family shows that Glu-2 is not a conserved residue, instead, there is a predominance in this position of non polar amino acids, and also deletions (21). Consequently, Glu-2 would not have any structural or functional relevance for these toxins.
In summary, St I E2C and St I R52C conserve the main conformational properties of the wild type and show similarities in binding to liposomal vesicles, whereas they differ in their permeabilizing activity. Therefore, St I E2C and St I R52C constitute good tools to study the steps of the permeabilizing mechanisms of sticholysins that take place after binding to the membrane, using thiol-specific probes such as fluorescent and spin labels.
REFERENCES
1. Lanio ME, Morera V, Álvarez C, Tejuca M, Gomez T, Pazos F, et al. Purification and characterization of two hemolysins from Stichodactyla helianthus. Toxicon. 2001;39:187-94.
2. Tejuca M, Serra MD, Ferreras M, Lanio ME, Menestrina G. Mechanism of membrane permeabilization by Sticholysin I, a cytolysin isolated from the venom of the sea anemone Stichodactyla helianthus. Biochemistry. 1996;35:14947-57.
3. Athanasiadis A, Anderluh G, Macek P, Turk D. Crystal structure of the soluble form of equinatoxin II, a pore-forming toxin from the sea anemone Actinia equina. Structure. 2001;9:341-6.
4. Hinds MG, Zhang W, Anderluh G, Hansen PE, Norton RS. Solution structure of the eukariotic pore-forming cytolisin equinatoxin II: implication for pore formation. J Mol Biol. 2002;315:1219-29.
5. Castrillo I, Alegre-Cebollada J, del Pozo AM, Gavilanes JG, Santoro J, Bruix M. 1H, 13C, and 15N NMR assignments of the actinoporin Sticholysin I. Biomol NMR Assign. 2009;3(1):5-7.
6. Mancheño JM, Martin-Benito J, Martínez-Ripoll M, Gavilanes JG, Hermoso JA. Crystal and electron microscopy structures of Sticholysin II actinoporin reveal insight into the mechanism of membrane pore formation. Structure. 2003;11:1319-28.
7. Kristan KC, Viero G, Dalla Serra M, Macek P, Anderluh G. Molecular mechanism of pore formation by actinoporins. Toxicon. 2009;54(8):1125-34.
8. Hong Q, Gutierrez-Aguirre I, Barlic A, Malovrh P, Kristan K, Podlesek Z, et al. Two-step membrane binding by Equinatoxin II, a pore-forming toxin from the sea anemone, involves an exposed aromatic cluster and a flexible helix. J Biol Chem. 2002;277:41916-24.
9. Bacrakč B, Gutiérrez-Aguirre I, Podlesek Z, Sonnen AFP, Gilbert RJ, Macek P, et al. Molecular determinants of sphingomyelin specificity of a eukaryotic pore forming toxin. J Biol Chem. 2008;283:18665-77.
10. Gutiérrez-Aguirre I, Barlic A, Podlesek Z, Macek P, Anderluh G, Gonzalez-Mañas JM. Membrane insertion of the N-terminal alpha-helix of equinatoxin II, a sea anemone cytolytic toxin. Biochem J. 2004;384:421-8.
11. Malovrh P, Viero G, Dalla Serra M, Podlesek Z, Lakey JH, Macek P, et al. A novel mechanism of pore formation: membrane penetration by the N-terminal amphipathic region of equinatoxin. J Biol Chem. 2003;27:22678-85.
12. Belmonte G, Pederzolli C, Macek P, Menestrina G. Pore formation by the sea anemone cytolysin equinatoxin II in red blood cells and model lipid membranes. J Membr Biol. 1993;131:11-22.
13. Alegre-Cebollada J, Martínez DP, Gavilanes JG, Goormaghtigh E. Infrared spectroscopy study on the conformational changes leading to pore formation of the toxin sticholysin II. Biophys J. 2007;93:3191-201.
14. Tejuca M, Anderluh G, Macek P, Marcet R, Torres D, Sarracent J, et al. Antiparasite activity of sea anemone cytolysins on Giardia duodenalis and specific targeting with anti-giardia antibodies. Int J Parasitol. 1999;29:489-98.
15. Panchal RG, Smart ML, Bowser DN, Williams DA, Petrou S. Pore-forming proteins and their applications in biotechnology. Curr Pharm Biotechnol. 2002;3:99-115.
16. Potrich C, Tomazzolli R, Dalla Serra M, Anderluh G, Malovrh P, Macek P, et al. Cytotoxic activity of a tumor protease-activated pore/forming toxin. Bioconjug Chem. 2005;16:369-76.
17. Tejuca M, Díaz I, Figueredo R, Roque L, Pazos F, Martinez D, et al. Construction of an immunotoxin with the pore forming protein StI and ior C5, a monoclonal antibody against a colon cancer cell line. Int Immunopharmacol. 2004;4:731-44.
18. Liu S, Wang H, Currie BM, Molinolo A, Leung HJ, Moayeri M, et al. Matrix metalloproteinase-activated anthrax lethal toxin demonstrates high potency in targeting tumor vasculature. J Biol Chem. 2008;283:529-40.
19. Braha O, Walker B, Cheley S, Kasianowicz JJ, Song L, Gouaux JE, et al. Designed protein pores as components for biosensors. Chem Biol. 1997;4(7):497-505.
20. Astier Y, Bayley H, Howorka S. Protein components for nanodevices. Curr Opin Chem Biol. 2005;9:576-84.
21. Kristan K, Viero G, Macek P, Dalla Serra M, Anderluh G. The equinatoxin N-terminus is transferred across planar lipid membranes and helps to stabilize the transmembrane pore. FEBS J. 2007;274:539-50.
22. Álvarez C, Mancheño JM, Martínez D, Tejuca M, Pazos F, Lanio ME. Sticholysins, two pore-forming toxins produced by the caribbean sea anemone Stichodactyla helianthus: their interaction with membranes. Toxicon. 2009;54(8):1135-47.
23. Pazos F, Valle A, Martinez D, Ramirez A, Calderon L, Pupo A, et al. Structural and functional characterization of a recombinant sticholysin I (rSt I) from the sea anemone Stichodactyla helianthus. Toxicon. 2006;48:1083-94.
24. Laemmli UK. Cleavage of structural protein during assembling of the head of bacteriophage T4. Nature. 1970;227: 680-5.
25. Martínez D, Campos AM, Pazos F, Alvarez C, Lanio ME, Casallanovo F, et al. Properties of St I and St II, two isotoxins isolated from Stichodactyla helianthus: a comparison. Toxicon. 2001;39(10):1547-60.
26. Rouser G, Fkeischer S, Yamamoto A. Two dimensional thin layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids. 1970; 5(5):494-6.
27. Lakowicz, JR. Principles of Fluorescence Spectroscopy. 2nd ed. New York: Kluwer Academic Publishers -Plenum Press; 1999.
28. Huerta V, Morera V, Guanche Y, Chinea G, González LJ, Betancourt L, et al. Primary structure of two cytolysin isoforms from Stichodactyla helianthus differing in their hemolytic activity. Toxicon. 2001;39: 1253-6.
29. Gromiha MM, An J, Kono H, Oobatake M, Uedaira H, Sarai A. ProTherm: Thermodynamic Database for Proteins and Mutants. Nucleic Acids Res. 1999;27(1): 286-8.
30. Venyaminov SY, Yang JT. Chapter 3: Determination of protein secondary structure. In: Fasman GD, editor. Circular Dichroism and the Conformational Analysis of Biomolecules. New York: Plenum Press; 1996. p. 109-82.
31. Kelly SM, Jess TJ, Price NC. How to study proteins by circular dichroism. Biochim Biophys Acta. 2005;1751:119-39.
32. Kristan K, Podlesek Z, Hojnik V, Gutierrez-Aguirre I, Gunchar G, Turk D, et al. Pore formation by Equinatoxin II, a eukaryotic pore-forming toxin, requires a flexible N-terminal and a stable b-sandwich. J Biol Chem. 2004;279:46509-17.
33. Anderluh G, Pungercar J, Krizaj I, Strukelj B, Gubensek F, Macek P. N-terminal truncation mutagenesis of equinatoxin II, a pore-forming protein from the sea anemone Actinia equina. Protein Eng. 1997;10:751-5.
34. Wang Y, Lee Chua K, Eng Khoo H. A new cytolysin from the sea anemone, Heteractis magnifica: isolation, cDNA cloning and functional expression. Biochim BiophysActa. 2000;1478:9-18.
35. Pazos IF, Martínez D, Tejuca M, Valle A, del Pozo A, Álvarez C, et al. Comparison of pore-forming ability in membranes of a native and a recombinant variant of Sticholysin II from Stichodactyla helianthus. Toxicon. 2003;42:571-8.
36. Castrillo I, Araujo NA, Alegre-Cebollada J, Gavilanes JG, Martínez-del-Pozo A, Bruix M. Specific interactions of sticholysin I with model membranes: An NMR study. Proteins. 2010;78:1959-70.
37. Anderluh G, Barlic A, Podlesek Z, Macek P, Pungercar J, Zecchini ML, et al. Cysteine-scanning mutagenesis of a eukariotic pore-forming toxin from sea anemone. Eur J Biochem. 1999; 263:128-36.
38. Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis. 1997;18:2714-23.
Received in May, 2010.
Accepted for publication in March, 2011.

{kind=link}
{kind=link}
{kind=link}