










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBiotecnología Aplicada
versión On-line ISSN 1027-2852
Biotecnol Apl v.28 n.1 La Habana ene.-mar. 2011
RESEARCH
Comparison of three methods for DNA extraction from paraffin-embedded tissues
Comparación de tres métodos de extracción de ADN de tejidos embebidos en parafina
Yaxsier de Armas1, Virginia Capó1, Ledy X López1, Lilian Mederos2, Raúl Díaz2
1 Departamento de Anatomía Patológica
2 Laboratorio Nacional de Referencia e Investigaciones de Tuberculosis y Micobacterias, Instituto de Medicina Tropical Pedro Kourí, IPK Autopista Novia del Mediodía, Km 6½, CP 601, Marianao 13, La Habana, Cuba
ABSTRACT
DNA extraction from paraffin-embedded tissue (PET) is a critical step for many molecular techniques. Several protocols have been carried out for this objective according to the literature. In the present study, the performances of three DNA extraction methods from PET were compared to establish the optimal protocol for our laboratory. Ten lymph nodes from ten patients dying of AIDS were investigated. Histological and bacteriological studies were performed in lymph nodes samples. DNA was extracted using three methods: boiling for 20 minutes in distilled water (Method A); boiling for 30 minutes in 5% Chelex-100 resin solution (Method B) and a 4-hours lasting proteinase K digestion (Method C). PCR with specific sequence (IS 6110) were evaluated for the identification of Mycobacterium tuberculosis in PET. The DNA extract by the three methods was degraded. Statistical differences were observed when three DNA extraction methods were compared according to the purity of extracted DNA. Only with Methods B and C successful amplification was obtained. The last method (C) was the more time consuming of all. These results demonstrated that the Chelex-100 DNA extraction method (Method B), which uses a quelating resin, is useful as a routine method to achieve DNA extraction with good enough quality and quantity in a short period of time from PET. Method B is a good option in molecular pathology research.
Keywords: DNA extraction, paraffin, Mycobacterium tuberculosis, molecular pathology.
RESUMEN
La extracción de ADN de tejidos embebidos en parafina (TEP) es crucial en muchos estudios moleculares. Varios han sido los protocolos utilizados para conseguir tal propósito. En esta investigación se compararon tres métodos de extracción de ADN con la finalidad de seleccionar uno para el trabajo en nuestro laboratorio. Se tomaron diez ganglios linfáticos de diez fallecidos por sida, para su estudio histológico y bacteriológico. El ADN se extrajo por calentamiento a 100 °C (método A); con el empleo de resina quelante Chelex-100 (método B) y por digestión con proteinasa K (método C). Para la identificación de Mycobacterium tuberculosis se amplificó una región de la secuencia específica de inserción IS 6110, mediante la reacción en cadena de la polimerasa. El ADN de los TEP se obtuvo degradado, con diferencias significativas de pureza. Solo se logró amplificación con los métodos B y C; este último fue el más laborioso. El método que requirió resina quelante Chelex-100 (método B) fue el más útil: se obtuvo ADN con calidad y cantidad suficiente en un menor tiempo. Por tanto, este método puede ser considerado como una buena opción en patología molecular.
Palabras clave: extracción de ADN, parafina, Mycobacterium tuberculosis, patología molecular.
INTRODUCTION
The massive collections of paraffin-embedded tissue (PET) samples amassed by most Pathological Anatomy laboratories have become a valuable source for molecular pathology studies (1). However, although the tissue in PET samples has undergone fixation with formaldehyde and embedment in paraffin to preserve its structure, extracting DNA from such samples with enough quality for further molecular biology techniques has proven troublesome (2).
Several different methods tackling this problem have been published during the last decade. Some are based on proteolytic treatments for varying periods of time (3), sometimes followed by the use of organic solvents such as phenol and chloroform before precipitating the sample with ethanol (4); whereas other methodologies heat the samples in distilled water to lyse the cells (5). A method using Chelex-100 has been used with relative success (6), and some protocols sonicate the sample and employ treatments with detergents (7). Even commercial kits have been recently developed for this purpose (8).
The present work compared three methods for the extraction of DNA from PET samples, in order to select a single protocol based on considerations of DNA yield, ease of manipulation and speed. The best method would be used to complement the diagnosis of Mycobacterium tuberculosis in the Pathological Anatomy department at Pedro Kourí Tropical Medicine Institute (IPK).
MATERIALS AND METHODS
Sample processing
A lymph node was selected and extracted from each of the ten Acquired Immunodeficiency Syndrome (AIDS) patients deceased at the hospital for the attention of AIDS/Human Immunodeficiency Virus (HIV) patients, at IPK in Havana. The macroscopic appearance of each lymph node was recorded and samples taken for the histological and bacteriological study.
One half from each lymph node was embedded in paraffin for the histological detection of acid-alcohol resistant bacilli (AARB), and the other half was sent to the National Reference Laboratory for Research on Tuberculosis and Mycobacteria at IPK (LNRI-TB) for the isolation of mycobacteria.
Three methods, denominated A, B and C, were selected for DNA extraction. Fifteen 10-µm histological sections were then performed from each of the ten paraffin-embedded lymph nodes. The sections were evenly distributed into three 1.5 mL microtubes (five per tube) for each individual sample.
DNA extraction
Method A
The sample was washed once with xylene and then twice with ethanol, as described by Lench et al. (5), followed by resuspension into 300 µL of sterile distilled water and and boiled for 20 min. It was then centrifuged at 13 000 rpm for 10 min, transferring the resulting supernatant to a fresh 1.5 mL microtube.
Method B
The tissue sections were directly immersed into 300 µL of a 5% Chelex-100 (Sigma, USA) suspension and heated at 100 ºC for 30 min, as described by Van den Zarden et al. (9). They were then centrifuged at 13 000 rpm for 10 min, transferring the resulting supernatant to a sterile 1.5 mL microtube. Care was taken during transfer to prevent resin carryover.
Method C
The tissue sections were washed once with xylene and twice with ethanol, similarly to method A. They were then digested by resuspension into 300 µL of TEN buffer (Tris-HCl 0.04 M, pH 8.3; NaCl 0.2 M, pH 8.0; EDTA 1 mM) containing 5 µL of Proteinase K (Merck, Darmstadt, Germany, 20 mg/mL) and 5.25 µL of 20% SDS as described by Ghossein et al. (10). After incubation at 55 ºC for 4 hours the sample was heated at 100 ºC for 10 min to inactivate Proteinase K and centrifuged at 13 000 rpm for 10 min. The resulting supernatant was precipitated by adding two volumes of absolute ethanol in the presence of NaCl (0.2 M final concentration), incubating at room temperature, and centrifuging again at 13 000 rpm for 30 min. The precipitate was dissolved in 40 µL of sterile distilled H2O.
Polymerase Chain Reaction
The quality of the DNA produced by the three extraction methods was assessed through the amplification by polymerase chain reaction (PCR) of a 245 bp fragment from the M. tuberculosis IS 6110 insertion sequence (11). The reaction mixtures (50 µL) contained 10 mM Tris/HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 200 µM deoxyribonucleotide tri-phosphates (dNTP), 0.4 mM each of primers INS-1 and INS-2, 2 units of Taq DNA polymerase (Bioline, UK) and 5 µL of DNA template, extracted by each of the three methods. The amplification profile used was 96 ºC for 3 min, 30 cycles of 1 min at 96 ºC, 1 min at 65 ºC and 2 min at 72 ºC, and a final extension step of 6 min at 72 ºC. Amplification of the β-globin gene was used as an internal experimental control to correct for the presence of PCR inhibitors in the extracts, as described by Schewe et al. (12). The PCR products were resolved on 1.2% agarose gels, visualized by ethidium bromide staining, and photographed under UV trans-illumination with a Power Shot G6 digital camera (Canon, Japan).
As a positive control, all the PCR reaction runs employed DNA extracted from a strain of M. tuberculosis (H37Rv), provided by LNRI- TB. Sterile distilled water in place of the DNA template was used as negative control. Three PCR replicates were run per sample.
Statistical analysis
The data were analyzed with the software application Statistical Package for the Social Sciences (SPSS) for Windows (version 11.5). The Kruskal-Wallis test was used to compare the means from optical density (OD) ratios at 260 and 280 nm (OD260/OD280). Ten OD measurements were made per DNA extraction method.
RESULTS
All three methods produced only degraded DNA (Figure 1), although the extent of degradation was variable. Method A produced the largest degradation, whereas method C produced the least degraded DNA. Method B yielded roughly intermediate results.
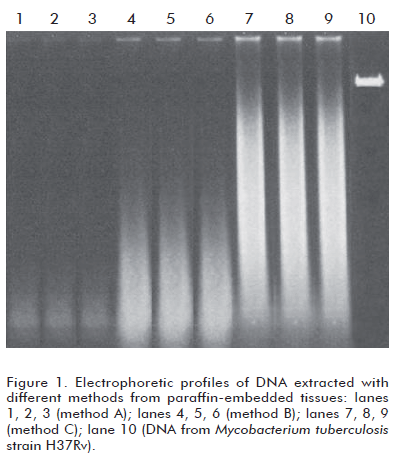
The mean OD260/OD280 ratios were 1.33 (method A), 1.63 (method B) and 1.67 (method A). Method A resulted in some statistical differences from methods B and C (p = 0.023), and no differences were detectable between the latter ones (p = 0.073).
In order to evaluate the quality of the extracted DNA, we attempted to amplify the M. tuberculosis IS 6110 insertion sequence. A successful amplification was only possible with template obtained from methods B and C (Figure 2). No inhibitors of the PCR reaction were present in extracts prepared by any of the three methods, as the β-globin gene was readily amplified in all cases.
In terms of speed, method B was the least laborious, requiring only 8 min for its completion. Method C, on the other hand, requires multiple steps and a longer time (20 min) than methods A and B. Method A requires 15 min.
The results of the amplification of the IS 6110 insertion sequence from the PET samples, that is, the identity of positive and negative samples, matched those obtained by conventional bacteriological tests at the LNRI -TB.
DISCUSSION
This is the first report on molecular detection of M. tuberculosis in PET samples, using three different DNA extraction methods, in Cuba.
DNA extraction is the first step for the application of many molecular techniques. Though apparently straightforward, many experimental failures can be traced back to problems in this stage that result in insufficient yields, poor DNA quality, contaminants at unacceptable levels and partial degradations; in addition, a poor choice of extraction method can unnecessarily lengthen a protocol (13). Trying to obtain high quality DNA from PET samples further compounds the issue; in fact, this has become the critical step in our own experimental runs (8).
In this study, the extent of degradation of the genetic material depended on the extraction method. Boiling the samples (method A) seems to play a role in obtaining extremely degraded DNA. The use of Chelex-100 (method B) blunts the detrimental effects of extreme heat by chelating polyvalent ion metals that would otherwise catalyze the aqueous hydrolysis of DNA at high temperatures (14). Methods employing Proteinase K, such as method C, are the most efficient for obtaining high molecular weight DNA. Proteinase K breaks the linkages between nucleic acids and proteins of PET samples in a much milder manner, thus producing DNA fragments that have, in average, much larger average lengths (4).
OD260/OD280 ratios were used to estimate the sample purities obtained with each of the three methods. Their inspection revealed that methods B and C (1.63 and 1.67) produce relatively less contaminated DNA than method A (1.33). Although variations in sample quality and quantity can definitely influence the purity of the obtained DNA, this factor can be ruled out in this occasion, given that identical amounts (5 x 10 μm[50 μm]) and type of tissue were analyzed in each case. On the other hand, the extraction volumes, the use of reagents such as Proteinase K, the chosen incubation times and the specific techniques employed for concentrating DNA also influence the purity of the obtained nucleic acids (15), and may help to explain why method C had the best OD ratio and the lowest amount of contaminants.
Failure to amplify by PCR a desired target sequence from a specific sample can sometimes be caused by average fragment lengths falling way below the size of the desired amplicon; a situation that is common in extremely degraded material (12). Such a situation arose with extracts obtained with method A, from which it was not possible to amplify the target insertion sequence (IS 6110). Similar results were published by Stein et al. when using this procedure to detect Coxiella burnetti in PET samples (16). Another possibility would be the total absence of the target sequence due to the degradation produced by heating the tissue. Although the presence of PCR inhibitors constitutes another possible explanation, it was possible, however, to amplify satisfactorily an internal control (β-globin gene) from these samples. DNA extracted by methods B and C were useful to identify M. tuberculosis in the analyzed tissues; both techniques have been successfully used before for this purpose (6, 9, 10, 17). Chelex-100 has received wide use for extracting DNA from PET samples (11, 18, 19). In a previous study, our group managed to amplify short DNA fragments from M. tuberculosis using this chelating resin (6). Method C, on the other hand, has also been successfully employed by several German laboratories to validate PCR assays for the detection of M. tuberculosis in PET (12).
Obtaining DNA from PET samples with satisfactory yield and purity in the shortest possible time is a critical requirement for many molecular pathology studies. Applying slow, costly or complicated multi-step methods on large-scale studies or clinical practice may become impractical (20), and such is precisely the main drawback of method C. Method B, on the other hand, is fast, safe, cheap, and allowed the amplification of the desired target sequence in every case. It produced DNA of satisfactory quality on a single step, with the added benefit of minimizing the chance of cross-contamination (another inconvenience of multi-step protocols such as method C).
There is a large demand for fast and reliable molecular methods for the detection of M. tuberculosis in PET samples among molecular pathology laboratories, given that i) the samples shipped to such facilities have generally been fixed in formaldehyde; preventing therefore the application of methods based on culturing the microorganism and limiting diagnostic options to the detection of AARB and histopathological examination; ii) the tissue alterations on which histopathological diagnosis is based are actually unspecific, as several granulomatous disorders produce tissue morphologies similar to those of tuberculosis; iii) microscopy techniques have a low sensitivity that becomes even lower in PET samples from HIV/AIDS patients with extra-pulmonary tuberculosis; iv) should a histopathological diagnosis of AARB-positive granulomatous lymphadenitis be reached, it would not be necessary to surgically obtain another lymph node sample for microbiological culture; and v) accurately identifying the microorganism responsible for the infection would allow a more targeted drug regime; a necessity arising out of the differential susceptibility to these agents exhibited by different mycobacterial species (21).
The results presented here demonstrate the usefulness of methods B and C for extracting DNA from PET samples to diagnose the presence of M. tuberculosis by PCR. However, given the working conditions and the characteristics of the patients attending the hospital at IPK, method B is recommended as the most appropriate choice for the Pathological Anatomy laboratory due to the satisfactory compromise it provides between DNA quality and yields on one side and processing time on the other.
REFERENCES
1. García P, Benavente F, Melo A, Roa I, Roa JC. Efecto de la fijación en la calidad del ADN: estudio controlado con cinco fijadores. Rev Esp Patol. 2006;39:175-9.
2. Srinivasan M, Sedmak D, Jewell S. Effect of fixatives and tissue processing on the content and integrity nucleic acids. Am J Pathol. 2002;161:1961-71.
3. Jackson DP, Lewis FA, Taylor GR, Boylston AW, Quirke P. Tissue extraction of DNA and RNA and analysis by the polymerase chain reaction. J Clin Pathol. 1990;43:499-504.
4. Wright DK, Manos MM. Sample preparation from paraffin-embedded tissues. En: Innis MA, editor. PCR protocols: a guide to methods and applications. San Diego: Academic Press; 1990. p. 153-6.
5. Lench N, Stainer P, Williamson R. Simple non-invasive method to obtain DNA for gene analysis. Lancet. 1988; 1(8599):1356-8.
6. de Armas Y, Capó V, González E, Mederos LM, Díaz R. Extracción de ADN de tejidos embebidos en parafina por Chelex-100. Rev Esp Patol. 2006;39:171-4.
7. Heller MJ, Robinson RA, Burgart U, TenEyck CJ, Wilke WW. DNA extraction by sonication: a comparison of fresh, frozen, and paraffin-embedded tissues extracted for use in polymerase chain reaction assays. Mod Pathol. 1992;5:203-6.
8. Hellmann A, Rohleder U, Schmitter H, Wittig M. STR typing of human telogen hairs- a new approach. Int J Legal Med. 2001;114:269-73.
9. Van der Zanden AG, Hoentjen AH, Heilmann FG, Weltevreden EF, Schouls LM, van Embden JD. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis complex in paraffin wax embedded tissues and in stained microscopic preparations. Mol Pathol. 1998;51:209-14.
10. Ghossein RA, Ross DG, Salomon RN, Rabson AR. A search for mycobacterial DNA in sarcoidosis using the polymerase chain reaction. Am J Clin Pathol. 1994; 101:733-7.
11. van Embden JDA, Cave MD, Crawford JT, Dale JW, Eisenach KD, Gicquel B, et al. Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: recommendations for a standardized methodology. J Clin Microbiol. 1993;31:406-9.
12. Schewe C, Goldmann T, Grosser M, Zink A, Schlüns K, Pahl S, et al. Inter-laboratory validation of PCR-based detection of Mycobacterium tuberculosis in formalin-fixed, paraffin-embedded tissues. Virchows Arch. 2005;447:573-85.
13. De Armas Y, Rodríguez MM, Bisset JA, Fraga J. Modificación de un método de extracción de ADN genómico de Aedes aegypti (Diptera: Culicidae). Rev Colomb Entomol. 2005;31:203-6.
14. Singer-Sam J, Tanguay RL, Riggs AD. Use of chelex to improve the PCR signal from small number of cells. Amplifications. 1989;3:11.
15. Bonin S, Petrera F, Rosai J, Stanta G. DNA and RNA obtained from Bouin’s fixed tissues. J Clin Pathol. 2005;58:313-6.
16. Stein A, Raoult. A simple method for amplification of DNA from paraffin-embedded tissues. Nucleic Acids Res. 1992;20:5237-8.
17. Popper HH, Klemen H, Hoefler G, Winter E. Presence of mycobacterial DNA in sarcoidosis. Human Pathol. 1997;28:796-800.
18. Romero RL, Juston AC, Ballantyne J, Henry BE. The applicability of formalin-fixed and formalin fixed paraffin embedded tissues in forensic DNA analysis. J Forensic Sci. 1997;42:708-14.
19. Pak F, Pyakural P, Kokhaei P, Kaaya E, Pourfathollah AA, Selivanova G, et al. HHV-8/KSHV during the development of Kaposi’s sarcoma: evaluation by polymerase chain reaction and immunohistochemistry. J Cutan Pathol. 2005;32:21-7.
20. García LA, Rodrigo JP, Sánchez P, Ramos S, Suárez C. Extracción de ADN con resina chelex en el análisis de la amplificación oncogénica en carcinomas de cabeza y cuello. Acta Otorrinolaringol Esp. 2004;55:139-44.
21. Krishna K, Singh M, Muralidhar M, Kumar A, Chattopadhyaya TK, Kapila K, et al. Comparison of in house polymerase chain reaction with conventional techniques for the detection of Mycobacterium tuberculosis DNA in granulomatous lymphadenopathy. J Clin Pathol. 2000; 53:355-61.
Received in July, 2010.
Accepted for publication in April, 2011.
Yaxsier de Armas, Departamento de Anatomía Patológica, Instituto de Medicina Tropical Pedro Kourí, IPK Autopista Novia del Mediodía, Km 6½, CP 601, Marianao 13, La Habana, Cuba, E-mail: yaxsier@ipk.sld.cu

{kind=link}