










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBiotecnología Aplicada
versión On-line ISSN 1027-2852
Biotecnol Apl vol.28 no.3 La Habana jul.-sep. 2011
TECHNIQUE
Quick transfer of Open Reading Frames between Yeast two hybrids and Protein fragment complementation assay vectors by homologous recombination cloning in bacteria
Transferencia rápida de los marcos abiertos de lectura entre vectores del sistema de dos híbridos de levadura y vectores de ensayo de complementación de proteínas mediante clonaje por recombinación homóloga en bacterias
Yelaine Tejeda, Julio Raúl Fernandez, Amanda B Colarte, Clara Y Taylor
División de Inmunodiagnóstico y Genómica, Centro de Ingeniería Genética y Biotecnología, CIGB. Ave. 31 e/ 158 y 190, PO Box 6162, CP 10600, Playa, La Habana, Cuba.
ABSTRACT
A common rate-limiting step in many high throughput proteome scale analyses is the cloning of predicted open reading frames (ORFs) into technique-specific vectors. For example, methodologies for the detection of protein interactions such as Yeast-two-hybrid (Y2H) assay require validation using alternative methods as Protein Fragment Complementation Assays (PCA) or pulldown experiments. Various experimental alternatives for rapid homologous recombination gene transfer in vivo between the Y2H and PCA vectors were evaluated. Two sets of universal primers sharing an overlap of 31 b homology between inserts and acceptor vectors were designed and PCR performance conditions were tested. Cotransformation of PCR products with the digested acceptor vector in E. coli strain allowed homologous recombination cloning. The method was proved to be effective for cloning 5 ORFs with sizes ranging from 0.294 to 1.2 kb. The proposed method allows the quick transfer of any open reading frames between the Y2H and PCA assay vector systems by using a universal set of primers. It doesn’t depend on the presence of specific restriction sites in the acceptor vector or causes changes in the open reading frame. This system will be useful for routine validation of protein interactions. We also report here the feasibility of DH10B strain for homologous recombination cloning.
Keywords: Homologous recombination cloning, DH10B, high throughput, Protein fragments complementation assay, yeast-two-hybrid assay.
RESUMEN
Un paso limitante en muchos análisis del proteoma a alto flujo es la clonación de losmarcos abiertos de lectura (ORF) en los vectores específicos para cada técnica. Por ejemplo, las metodologías para la detección de interacciones entre proteínas tales como el ensayo de dos híbridos de levadura (Y2H) requieren una validación utilizando métodos alternativos, como por ejemplo los ensayos de complementación de fragmentos de la proteína (PCA) o experimentos de precipitación de proteinas. Diversas alternativas experimentales para una rápida transferencia mediante la recombinación homóloga de genes in vivo entre los vectores Y2H y PCA fueron evaluadas. Dos juegos de cebadores universales con una superposición de homología entre 31 b de los insertos y los vectores aceptadores fueron diseñados y las condiciones de amplificación de la reacción de PCR fueron evaluadas. Los productos de PCR fueron cotransformados con el vector aceptor en cepas de E. coli que permiten la clonación mediante recombinación homóloga. El método demostró ser eficaz para la clonación de cinco ORFs con tamaños que oscilan entre 0.294 a 1.2 kb. El método propuesto permite la rápida transferencia de los marcos abiertos de lectura entre los vectores de los sistemas de Y2H y PCA mediante el uso de un conjunto universal de los cebadores. El método de clonaje no depende de la presencia de determinadas sitios de restricción en el vector aceptor y no provoca cambios en el marco abierto de lectura. Este sistema será útil para la validación de rutina de las interacciones de proteínas. También mostramos la viabilidad de la cepa DH10B para la clonación por recombinación homóloga.
Palabras clave: Clonaje por recombinación homóloga, DH10B, alto flujo, ensayo de complementación de fragmentos de proteínas, ensayo de dos híbridos de levadura.
INTRODUCTION
The analysis of protein-protein interactions is an invaluable resource for better comprehension of cell-ular functions and in the present is the central axe of many biomedical investigations. This allows the identification of proteins of therapeutic interest and increases the amount and quality of information related to potencial drugs [1]. Some methodologies such as pull down techniques, Yeast-two-hybrids (Y2H) assay and Protein fragment complementation assay (PCA) have been developed for the evaluation of protein-protein interactions. Each of them has potentialities and disadvantages, so using at least two different assays is highly recommended. The Y2H provides a mean to answer questions regarding protein-protein interactions, but has limitations which restrict its use to certain interaction networks; furthermore they provide little information regarding interaction localization at the subcellular level. The development of PCA employing a fluorescent reporter such as a member of the green fluorescent protein (GFP) family has led to a new method of interaction with the advantages of a very low background signal coupled with rapid detection of protein-protein interactions in vivo, while also providing information regarding interaction compartmentalization, but with a limited throughput.
Sometimes the number of interactions to be evaluated demands high throughput cloning of open reading frames (ORFs) which can not be acomplished by traditional restriction enzyme dependent cloning methods. To face this challenge, several high throughput cloning methodologies have been stablished. They are quick and efficient; do not depend on the presence of specific restriction sites or T4 DNA ligase and which is the most important feature, allowing gene transfer among vectors from different expression systems [2]. Several high throughput commercial cloning systems are available but they are too expensive for many working groups. Nevertheless, Parrish and co-workers reported in 2004 the high throughput cloning of Campylobacter jejuni ORFs using in vivo homologous recombination in Escherichia coli [3]. This methodology is based in the recombinant possibilities of RecA+ strains. It just requires PCR amplification of inserts with oligonucleotides containing sequences complementary to the regions of donor vector flanking the ORF to be transferred and also a region homologous to the 5’ and 3’ ends of the linearized acceptor vector. PCR products and the acceptor vector are then co-transformed in a RecA+ E. coli strain for the in vivo production of recombinant clones.
With the aim of creating a ORFs transferring system among Y2H assay vectors and PCA mammalian vectors for routine protein-protein interactions validation, various experimental alternatives using restriction enzyme independent in vivo cloning were designed.
MATERIALS AND METHODS
Microbial strains and plasmids
The E. coli strain used were DH10B (F- mcrA, D (mrr-hsdRMS-mcrBC) f 80, lacZ DM15, D lacX74, deoR, recA1, araD39, D (ara-leu) 7697, galK, galU, rpsL, endA1, nupG) and BJ5183 (F - , endA, sbcB, recBC, galK, met, str R, thi-1, bioT, hsdR, l -). Competent cells of both strains, were obtained using polyethylene glycol and dimethyl sulfoxide PEG-DMSO [4] or CaCl2 [5] methods.
Plasmids pGBKT7, pGADT7 and pACT2 (Clontech, Palo Alto) from the Yeast two hybrids system were utilized as donor vectors whereas pcDNA3.1/Zeo (+) Venus (1)-Zipper and pcDNA3.1/Zeo (+) Venus (2)-Zipper [6] were used as acceptor vectors.
PCR amplification
Two sets of primers with different homology regions between donor and acceptor vectors were tested. The inserts from pGBKT7 were amplified using UT1 and UT2 oligonucleotides (Table 1). These oligonucleotides are complementary to the sequences of pGBKT7 flanking the ORF coding region and at the same time contain sequences homologous to the ends generated by pcDNA3.1/Zeo (+) Venus (1)-Zipper BspE I/Xba I digestion. The PCR generated products have a 16 b sequence homologous to the acceptor vector in its 5’ end as well as a 12 b homology sequence in the 3’ end. A second set of primers UT3 and UT4 was used to amplify the ORF sequence in order to increase the homology region to the acceptor vector up to 31 bp in both ends (Table 1).
The sequences from pGADT7 or pACT2 were amplified using UT5 and UT6 oligonucleotides (Table 1). These oligonucleotides are 31 bp complementary to the sequences of donor vectors flanking the ORF coding region and at the same time the oligonucleotides contain a 31 bp sequence homologous to the ends generated by pcDNA3.1/Zeo (+) Venus (2)-Zipper BspE I/Xba I digestion.
PCR amplifications from plasmid templates, containing 1 ng of plasmid and 300 nM of each PCR primer (94 ºC for 30 s, 60ºC for 30 s, and 72 ºC for 1:30 min for 30 cycles), were conducted using PCR master mix from Promega, according to the manufacturers’ instructions. All PCR products were purified with a QIAquick PCR Purification Kit from Qiagen. The size and purity of the expected products were verified by agarose electrophoresis.
In vivo cloning by homologous recombination
The linearized vector and the PCR amplified products were mixed in 50 µL of KCM (0.1 M KCl, 30 mM CaCl2, 50 mM MgCl2) and transformed in 50 µL of PEG-DMSO competent cells [4]. The following ng insert/ng vector ratios were assayed: 80:60, 50:20, 150:20, and 50:50. The mix was incubated for 10 min. LB medium (200 µL) was added, mixed, and incubated for 1 h at 37 °C. The complete transformation mix was plated on LB ampicillin culture plates. The plates were then incubated overnight at 37 °C.
Alternatively, thermal shock transformation was employed. To accomplish these goal fifty nanograms of acceptor vector and fifty nanograms of insert in 50 µL water were added to 50 µL CaCl2 quimiocompetent cells. The complete transformation mix was plated on LB ampicillin culture plates. The plates were then incubated overnight at 37 °C.
Analysis of recombinant clones
Single colonies from each construction were picked into 5 mL of LB ampicillin and incubated overnight at 37 °C. The DNA from clones was isolated using a QIAprep Miniprep Kits. The analysis of recombinant clones from pcDNA3.1 /Zeo (+) Venus 1 was performed by enzymatic digestion with Not I (Promega, Madison, WI) according to the manufacturer and also by PCR amplification with UT3 and UT4 oligonucleotides using PCR Master Mix (Promega, Madison, WI). In the case of pcDNA3.1 /Zeo (+) Venus 2 recombinant clones, DNA with a larger size that a vector on agarose gels were selected as positive. From each construction a representative clone was used for PCR confirmation using the UT5/UT6 oligonucleotide pair. DNA agarose gel electrophoresis (0.8%) was performed for the analysis of DNA digestions and PCR amplification.
RESULTS
Cells transformed with PCR products containing 12 bp and 16 bp homology tags did not yield colony growth in DH10B or BJ5183 using any of the transformation protocols. To enlarge the region of homology between the PCR and the acceptor vector, a second round of amplification using the oligonulceotides UT3 and UT4 was performed. Cloning of DNA fragments from pGBKT7 with 31 homology bases to pcDNA3.1/Zeo (+) Venus (1)-Zipper BspEI/XbaI produced the growth of 9 colonies in DH10B PEG-DMSO competent cells after transformation. No digestion analysis confirmed the obtention of 3 recombinant clones producing the expected electrophoretic pattern: a 294 bp fragment corresponding to the insert and a 5166 kb fragment corresponding to the acceptor vector’s size (Figure 1). The cloning efficiency was 33.3%. In all three cases the fragment insertion was confirmed by PCR using UT3 and UT4 oligonucleotides (Figure 2). The highest cloning efficiency was obtained using a 50:50 ng insert/ng vector ratio.
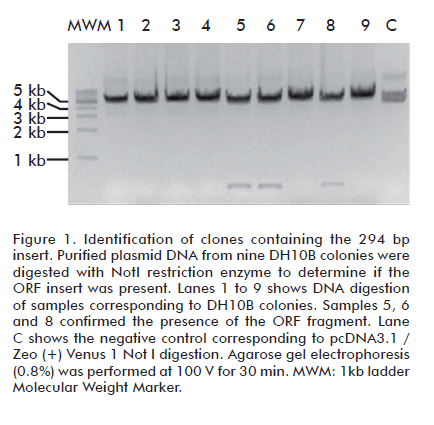

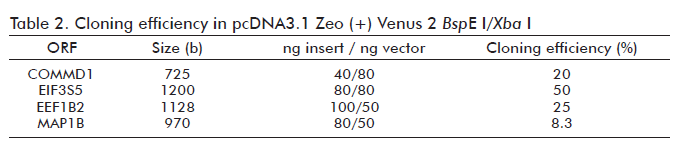
CaCl2 transformation did not yield any colony growth in DH10B. In addition, neither transformation protocols produce any colony when BJ5183 strain was used. This result was also obtained for ORFs transferred from pGADT7 or pACT2 to pcDNA3.1 Zeo (+) Venus 2. Table 2 shows the best ng insert/ng vector ratio for cloning purposes. A random selected positive clone from each construction was tested by PCR using oligonuclotides UT5 and UT6. The amplified product from tested clones showed the spected sizes of 0.725 kb for COMMD1, 0.970 kb for MAP1B, 1.128 kb for EEF1B2 and 1.2 kb for EIF3S5 (Figure 3). The results shown in Table 2 indicate that the in vivo recombination procedure succeeded, as all co-transformations led to the production of recombinant plasmids.

DISCUSSION
To make in vivo recombination cloning a feasible practice for a large number of ORF, the length of the identical sequences for recombination must be minimized to reduce the expense of generating primers for a large number of ORFs. A key problem to achieve this goal is that the efficiency of cloning has been shown to decrease as the length of homology between the vector and insert is decreased [7]. Otherwise, Bubeck, Winkler and Bautsch reported that about 10 bp overlap represented a practical minimum length to obtain sufficiently high numbers of transformants in DH5α [8]. In our conditions, we could not reproduce the results of Bubeck, Winkler and Bautsch. Co-transforming E. coli strains with the PCR products containing 16 bases 5’ recombination tag and 12 bases 3’ recombination tag did not yield positive clones. An increase in the region of homology up to 31 b results in a range of 8.3% to 50% efficiency of cloning by in vivo recombination. This confirms the findings of Parrish’s that points out the requirement of minimal 21 bp homology regions shared by vector and insert [3]. On the other hand our results are also in accordance with Oliner [7] who reported a recombination efficiency of 20% when the residues shared between vector and the PCR product is 30 bp in each end.
According to Parrish and co-workers there is variability in cloning efficiency among E. coli strains. They strongly recommend KC8 strain which allowed a 75% cloning efficiency, and also BUN10 (recB21 recC22 sbcA23), V324 (recD1009), MG1655 or DH5α (RecA1), in this order. Besides, Kong, Yang and Geller [10] used BJ5183 E. coli strain for routine manipulation of relatively large DNAs such as the five cosmids that comprise helper virus-free HSV-1 packaging system. In this system, recombination cloning required overlap sizes from 251 bp to 18 kb with 500 bp to 5 kb preferred.
Two E. coli strains have been routinely used in our laboratory: BJ5183 and DH10B. The use of DH10B has not been described previously in this context. Both strains are succesfully employed for production of recombinant adenovirus and DNA purification respectively. The oligonucleotides described in the present work have smaller overlaps which might cause the lack of BJ5183 recombinants. On the other hand, homologous recombination cloning by means of co-transformation of the digested acceptor vector and inserts in DH10B strain proved to be effective in the cloning of 5 ORFs with sizes ranging from 0.294 to 1.2 kb. Although cloning efficiency for DH10B strain was lower than those reported in Parrish work, it has an advantage over KC8 strain as suitability for extended storage [3].
Finally, we have found that the PEG-DMSO method for preparation and transformation of competent cells was superior that the CaCl2 method. This superiority is independent from the transformation efficiency as we used competent cells with similar transformation efficiency. Other authors relied on CaCl2 transformation for DH5α, and were able to replicate the successful cloning in this strain, but until efficiency 25-fold lower than that obtained using electroporated JC867 strain [7]. On the other side, Parrish recommends the use of the PEG-DMSO as a particular effective way for high throughput cloning by transformation in 96-well plates [3].
As illustrated in this study, the presented in vivo homologous recombination cloning methodology is quick, efficient and cheap. The method requires no in vitro enzymatic steps except the PCR amplification using pairs of universal oligonucleotides that allows gene transfer between two different expression systems. This will allow the evaluation and confirmation by PCA of those protein-protein interactions detected by Yeast two hybrids assay regardless the ORF fragments to be transferred. We also described the feasibility of DH10B strain for homologous recombination cloning. The present method was further validated in our lab by the direct cloning of ORFs amplified from human cDNAs into the pACT2 vector.
ACKNOWLEDGEMENTS
Authors wish to thank the generous colaboration of Dr. Stephen W. Michnick from the Biochemistry Department of Montreal University who gently donated the pcDNA3.1 Zeo (+) Venus-Zipper PCA vector system.
REFERENCES
1. Michnick SW, Ear PH, Manderson EN, Remy I, Stefan E. Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat Rev Drug Discov. 2007;6(7):569-82.
2. Marsischky G, LaBaer J. Many paths to many clones: a comparative look at high-throughput cloning methods. Genome Res. 2004;14(10B):2020-8.
3. Parrish JR, Limjindaporn T, Hines JA, Liu J, Liu G, Finley RL Jr. High-throughput cloning of Campylobacter jejuni ORfs by in vivo recombination in Escherichia coli. J Proteome Res. 2004;3(3):582-6.
4. Walhout AJ, Temple GF, Brasch MA, Hartley JL, Lorson MA, van den Heuvel S, et al. GATEWAY recombinational cloning: application to the cloning of large numbers of open reading frames or ORFeomes. Methods Enzymol. 2000;328:575-92.
5. Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166(4):557-80.
6. Remy I, Montmarquette A, Michnick SW. PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nat Cell Biol. 2004;6(4):358-65.
7. Oliner JD, Kinzler KW, Vogelstein B. In vivo cloning of PCR products in E. coli. Nucl Acids Res. 1993;21(22):5192-7.
8. Bubeck P, Winkler M, Bautsch W. Rapid cloning by homologous recombination in vivo. Nucl Acids Res. 1993;21(15):3601-2.
9. Takahashi N, Yoshikura H, Kobayashi I. An Escherichia coli strain, BJ5183, that shows highly efficient conservative (two-progeny) DNA double-strand break repair of restriction breaks. Gene. 2003;303:89-97.
10. Kong Y, Yang T, Geller AI. An efficient in vivo recombination cloning procedure for modifying and combining HSV-1 cosmids. J Virol Methods. 1999;80(2):129-36.
Received in November 2010.
Accepted for publication in August 2011.
Yelaine Tejeda. División de Inmunodiagnóstico y Genómica, Centro de Ingeniería Genética y Biotecnología, CIGB. Ave. 31 e/ 158 y 190, PO Box 6162, CP 10600, Playa, La Habana, Cuba. E-mail: yelaine.tejeda@cigb.edu.cu .

{kind=link}
{kind=link}