










My SciELO
 Custom services
Custom servicesServices on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.28 no.4 La Habana Sept.-Dec. 2011
RESEARCH
Levansucrase activity but not fructan accumulation in transgenic lsdA-expressing sugarcane recovered by optimized microprojectile bombardment of embryogenic calli
Actividad levanasacarasa pero ausencia de acumulación de fructanos en plantas transgénicas de caña de azúcar obtenidas mediante un procedimiento optimizado de bombardeo de callos embriogénicos
Alexander Banguela1,2, Raisa Rodríguez1,2, Juan G Arrieta1, Carmen Menéndez1, Elizabeth Kairúz1,3, Luis E Trujillo1, Lázaro Hernández1
1Centro de Ingeniería Genética y Biotecnología, CIGB. AP 6162, La Habana, Cuba.
2Instituto de Investigaciones en Fruticultura Tropical, IIFT. La Habana, Cuba.
3Departamento de Biología, Universidad Central Marta Abreu de Las Villas, UCLV. Santa Clara, Villa Clara, Cuba.
ABSTRACT
Sugarcane (Saccharum spp. hybrid) emerges as an ideal crop for the cost-effective transgenic production of fructans due to its high efficiency for fixing carbon and storing the substrate sucrose. As other gramineous species, sugarcane is recalcitrant to genetic transformation. In this work, we optimized conditions for the transformation of sugarcane cv. C1051-73 via microprojectile bombardment of embryogenic calli. The genes encoding the enhanced green-fluorescent protein (eGFP) and the neomycin phosphotransferase (nptII), both under the control of the maize ubiquitin 1 (Ubi-1) promoter, were used for the early detection of transient transformation events and for the selection of stable transformants, respectively. DNA was efficiently delivered into the cell without causing drastic damages in calli bombarded at the distance of 11 cm and the argon pressure of 90 PSI. Non-mosaic transgenic plantlets were recovered by increasing the geneticin concentration from 20 mg/L during callus growth to 25 mg/L for the shooting and rooting steps. Moreover, using the optimized transformation procedure, we recovered twenty transgenic sugarcane lines carrying the diazotrophicus levansucrase gene (lsdA) modified for vacuolar targeting of the enzyme, as a strategy for fructan production. Southern blot and PCR analysis revealed the stable presence of the chimaeric in the primary stalk and sprouts of plants grown under field conditions. None of the transgenic lines accumulated levan in mature stems or leaves, although one of them showed evident levansucrase activity in leaf extracts.
Keywords: Sugarcane, genetic transformation, eGFP, levansucrase, fructan, levan, lsdA.
RESUMEN
La caña de azúcar (Saccharum spp. hybrid) es un cultivo ideal para la producción transgénica de fructanos a altos niveles debido a su marcada eficiencia en fijar el carbono atmosférico y almacenar sacarosa. Como otras gramíneas, es recalcitrante a la transformación genética. En este trabajo se optimizó la transformación de caña de azúcar cv. C1051-73 por la vía del bombardeo de callos embriogénicos. El empleo de los genes de la proteína verde fluorescente potenciada (eGFP) y la neomicina fosfotransferasa II (nptII), ambos bajo el control del promotor Ubi-1 de maíz, permitió la detección temprana de los eventos de transformación y la selección de transformantes estables, respectivamente. La entrada del ADN a la célula fue más eficiente en los callos bombardeados a 11 cm de distancia y presión de argón de 90 PSI, y no presentaron daños drásticos. El incremento de la concentración de geneticina de 20 mg/L en el estadio de callos, a 25 mg/L en los pasos de formación de brotes previno la generación de plantas falsas positivas o mosaicos. Con el objetivo de producir fructanos en caña de azúcar, se obtuvieron 20 líneas transgénicas portadoras del gen de la levanasacarasa de Gluconacetobacter diazotrophicus (lsdA) fusionado a señales de localización vacuolar. Experimentos de Southern blot y reacción en cadena de la polimerasa confirmaron la presencia del gen quimérico en el genoma de plantas crecidas y ahijadas en el campo. Ninguna planta acumuló levana en las hojas o los tallos maduros, a pesar la detección de actividad levanasacarasa en los extractos foliares de una de las líneas.
Palabras claves: Caña de azúcar, transformación genética, eGFP, levanasacarasa, fructano, levana, lsdA.
INTRODUCTION
Sugarcane (Saccharum spp. hybrids) is a highly polyploid plant widely cultivated in tropical and subtropical countries for sugar and alcohol production, animal feed, and other important applications. The plant stores high sucrose concentration in stems to reach an average carbohydrate yield of 10 ton / hectare / year [1]. In this sense, sugarcane emerges as an ideal crop for the transgenic conversion of the substrate sucrose into commercially attractive fructans by the action of microbial or plant fructosyltransferases.
The endophytic bacterium Gluconacetobacter diazotrophicus secretes a constitutively expressed levansucrase (LsdA, EC 2.4.1.10). The enzyme transfructosylation reaction on sucrose releases glucose while yielding the β-2,6-linked polyfructan levan with degree of polymerization (DP) above 104, in addition to the β-2,1-linked fructooligosaccharides (FOS) 1-kestose (G1↔2F1←2F) and nystose (G1↔2F1↔2F1←2F) [2]. Recombinant LsdA expressed in yeast kept the catalytic performance of the natural enzyme despite the occurrence of posttranslational modifications, such as glycosylation [3, 4]. Tobacco plants constitutively expressing a vacuolar-driven LsdA yielded high contents of polymerized levan in mature leaves and stem. FOS, however, were not detected in the transgenic organs despite the fact that leaf extracts did synthesize 1-kestose after sucrose incubation [5].
As many gramineous crop, sugarcane is recalcitrant to genetic transformation. Embryogenic callus is the most suitable target tissue for sugarcane transformation [6]. Though there are reports on gene transferring into sugarcane via Agrobacterium tumefaciens [7- 9], biolistics has been used more frequently [10]. In both methods, the efficiency of generating transgenic plants varies among the cane varieties.
Here, we show a procedure with conditions optimized for sugarcane cv. C1051-73 transformation by microprojectile bombardment using embryogenic calli as the starting material. The use of the enhanced green-fluorescent protein (eGFP) as a reporter marker allowed early detection of transient transformation events and the optimization of the bombardment conditions. The neomycin phosphotransferase gene (nptII) under the control of the maize ubiquitin 1 (Ubi-1) promoter [11] was successfully used to select stable, non-mosaic plants genetically modified for fructan production. Southern blot and PCR analysis confirmed the integration of the G. diazotrophicus levansucrase gene (lsdA) in the genome of field-grown plants, both in the primary stalk and the sprouts. LsdA activity was detected in leaf extracts but no levan accumulated in either leaves or mature stems of the transgenic lines.
MATERIALS AND METHODS
Plant material, genetic transformation and growth conditions
Plant tissue culture experiments were conducted on Medium H consisting of Murashige and Skoog [12] salts and vitamins, sucrose 20 g/L, coconut water 10% (v/v) and agar 6 g/L. The growth medium was supplemented with 2,4 dichlorophenoxy acetic acid (2,4 D) at 3 mg/L for calli growth or geneticin (G418) when needed. The incubation temperature was of 28°C. The lighting condition chose was of 75 µmoles/m/s of cool-daylight fluorescent 6200 K light at a 16:8 h light: dark photoperiod. Spindle sections were taken from 6-8 month old field-grown sugarcane cv. C1051-73 plants. The outer old leaf-base coverings were removed carefully; the inner segments were immersed in ethanol 70% (v/v) for 5 min and flamed for surface disinfection. After removal of the outer sheaths, the innermost undifferentiated tissue was cut into 2.5 cm-long pieces, placed on Medium H supplemented with 2,4 D, and kept in the dark for 8 weeks to obtain friable embryogenic calli. Various concentrations of the selection marker geneticin (0, 5, 10, 15, 20, 25, 50 mg/L) were tested to determine the minimal inhibitory concentrations for the following steps: calli growth on Medium H with 2,4 D in the dark, shoots formation in Medium H in the light, and plantlets growth to 3-cm high in Medium H in the light. Three replicates of each concentration were tested with 15 explants per replicate. Calli were bombarded with DNA-coated gold micro-projectiles using an inflow gun [13] with Argon pressure, 90 PSI; without pre-chamber; target distance, 11 cm; chamber vacuum, -28 PSI. Particles were prepared as described by Franks and Birch [14]. Following bombardment, calli were cultured for 2 days in Medium H with 2,4 D for cells recuperation and posteriorly divided into portions of approximately 3 mm in diameter, placed on Medium H with 2,4 D and geneticin 20 mg/L, and kept in the dark during 30 days with subculture at the 15th day. Actively growing calli were placed on Medium H with geneticin 25 mg/L and incubated for tissue differentiation under the above-mentioned lighting regime during 30 days. Regenerated shoots were individualized, transferred to fresh Medium H with geneticin 25 mg/L and kept in the light for rooting and growth during one extra month.
The geneticin-resistant plantlets were planted in pots containing an equal-rate mixture of soil and organic matter and grown under greenhouse conditions to the average height of 60 cm. Each plant was transferred to the field for sprouting and grown for one year until stems maturation. After harvesting, twenty selected lines were propagated for second vegetative generation.
Genetic constructs
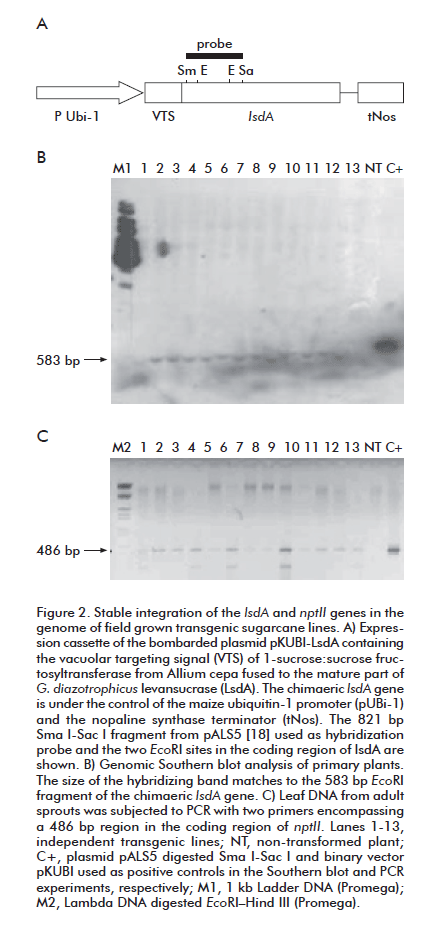
Standard methods were used for recombinant DNA procedures [15]. The binary vector pKUBI carrying the neomycin phosphotransferase II gene (nptII) as the selection marker was constructed by replacing the original CaMV 35S promoter from pCAMBIA 2300 (CAMBIA) by the maize Ubi-1 promoter from pUBI1, a derivative of pAHC25 [11]. Chimaeric levansucrase for plant vacuolar targeting was constructed by fusing the coding sequence for the mature part of Glucona-cetobacter diazotrophicus levansucrase (LsdA) to the first 219 nucleotides of the onion 1-sucrose:sucrose fructosyltransferase gene [5]. Plasmid pCMV2-EGFP (Invitrogene) was the source of the gene encoding the enhanced green fluorescent protein (eGFP) [16]. The coding region of eGFP and the chimaeric lsdA was placed under the transcriptional control of the constitutive Ubi-1 promoter and the nopaline-synthetase terminator (tNos). The expression cassettes were independently inserted in the HindIII site of the binary vector pKUBI to create the plasmids pKUBI-eGFP and pKUBI-LsdA.
Southern blot and PCR analysis of transgenic plants
Genomic DNA was extracted from 2 g of leaf tissue using the CTAB extraction method [17]. For Southern blot analysis, the genomic DNA (20 µg) was digested with EcoR I, electrophoresed through a 0.8% (w/v) agarose gel at 6 V/cm in TBE buffer, transferred to a nylon membrane (Hybond N, Amersham) by capillarity, and cross-linked to the membrane by UV. High-specific-activity DNA probe was generated by using a Prime-a-Gene labelling kit (Promega) with [α32P]dATP ( > 3000 Ci mmol-1; Amersham Pharmacia Bio tech) using as template the 821-bp SmaI-SacI fragment in the coding region of the lsdA gene in plasmid pALS5 [18]. Prehybridization for 4 h and hybridization for 16 h were performed at 65 °C in a solution of 7% (w/v) SDS, 1 mM EDTA, 1% (w/v) BSA in 10 mM sodium phosphate pH 7.2. High stringency washing was performed at 65 °C as follows: first wash in 2x SSC, 0.1% (w/v) SDS for 10 min; second and third washes in 1x SSC, 0.1% (w/v) SDS for 15 min, fourth and fifth washes in 0.1x SSC, 0.1% (w/v) SDS for 15 min. Membranes were then autoradiographed using Kodak X-OMAT X-K1 film at -70 oC, with intensifying screens.
The presence of the nptII gene in the transgenic lines was detected by PCR in 50-µL reactions containing undigested genomic DNA (100 ng) as template and the primers 5´AGACAATCGGCTGCTCTGAT 3´and 5´CATGTGTCACGACGAGATCC 3´. PCR conditions were 94 oC for 2 min, 35 cycles of cycling at 94 oC for 45 s, 55 oC for 45 sec and 72 oC for 1 min followed by incubation at 72 oC for 5 min.
LsdA activity and carbohydrate analysis in plant samples
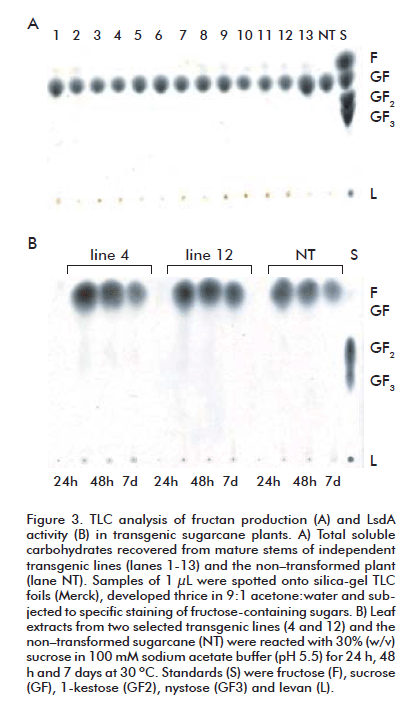
Levansucrase (LsdA) activity was assayed in leaf extracts. Leaf samples (5 g) were ground with liquid nitrogen in a mortar, and soluble proteins were recovered in 3 mL of the extraction solution consisting of 0.5 mM EDTA, 1 mM PMSF, 0.1% (w/v) TWEEN 20, 0.5% (w/v) soluble PVP, 50 mM Tris-HCl (pH 7.4) and 0.01% (w/v) NaAz. Cell debris was removed by centrifugation and the sample supernatant was reacted for 24 h, 48 h and one week with 30% (w/v) sucrose in 100 mM sodium acetate buffer (pH 5.5) at 30ºC.
Levan accumulation was assayed in mature leaves and stems. Plant leaf samples (3 g) were ground with liquid nitrogen in a mortar, and recovered in 3 mL of water. The homogenate was transferred to 15 mL corning tubes, mixed by vortex, and incubated for 15 min at 90 °C. After removal of cell debris by centrifugation at 10 000 × g for 15 min, the supernatant (designed as plant extract) was directly assayed for total fructan content. Stem slides were peeled out and squeezed for carbohydrate analysis. High-DP polysaccharides in cane juice and leaf extracts were 20x concentrated by ethanol (60% v/v) precipitation at -20 ºCand dissolved in deionised water.
Carbohydrates samples (1 mL of LsdA reaction products, plant extracts or concentrated polysaccharides) were spotted and separated by thin-layer chromatography (TLC) on silica-gel 60 plates (Merck KGaA, Darmstadt, Germany) using acetone-water (9:1) as the mobile phase. After three runs, the fructose-containing sugars were visualized by spraying the plates with a solution of 3% (w/v) urea, 1 M phosphoric acid in water-saturated butanol, and heating at 120 oC for about 5 min [19].
RESULTS AND DISCUSSION
Optimized conditions for genetic transformation of sugarcane cv. C1051-73 by microprojectile bombardment of embryogenic callus
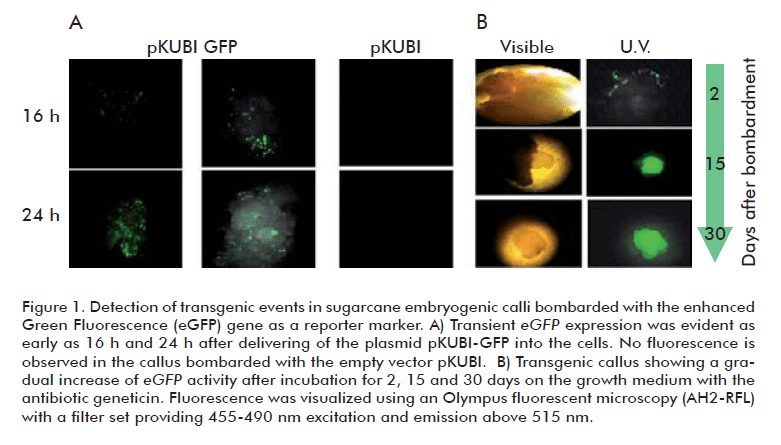
The green fluorescent protein (GFP) has been successfully used as a reporter for Agrobacterium-mediated transformation of sugarcane [20]. In this work, we used the enhanced GFP (eGFP) as the screening marker for early identification of transient transformation events in embryogenic calli subjected to microprojectile bombardment, aiming to establish an optimized transformation procedure for sugarcane cv. C1051-73. Conditions for efficient DNA delivery into the cells were established using the plasmid pKUBI-eGFP constructed for constitutive co–expression of the marker genes eGFP (reporter) and nptII (selection) in monocots. Fluorescence of eGFP in the transformed calli was observed as soon as 16 h after the bombardment (Figure 1A). The target distance and argon pressure for microprojectile bombardment were established to be optimal at 11 cm and 90 PSI, respectively. Under this condition, cell fluorescence was maximal while damages in the bombarded tissues remained to be rather slight. Other parameters such as chamber vacuum at -28 PSI, DNA concentration above 20 µg, and gold microparticules size below 6 µm were found to be important for efficient cell transformation, as it was previously reported [14]. The eGFP-positive calli grew without showing necrosis under geneticin selection, but failed to regenerate plantlets. As a fluorescent molecule, GFP is expected to generate free radicals upon excitation causing toxicity of the transformed plant cell [21]. Nevertheless, the early detection of the fluorescent cells and their successful multiplication in the presence of the selection antibiotic during callus growth (Figure 1B) supports the convenience of using eGFP as a reporter gene to monitor transient transformation events in sugarcane.
The sensitivity of sugarcane cv. C1051-73 to geneticin was examined in the range 0-50 mg/L at different stages during tissue culture. Calli reduced growth and became necrotic after 15 days in the presence of geneticin at 20 mg/L, while the minimal antibiotic concentration required for total inhibition of the shooting and rooting processes was 25 mg/L. Raza et al. [22] reported variations in the geneticin concentration required for regeneration inhibition of non-transformed calli in five sugarcane cultivars with values ranging between 25 and 60 mg/L.
The conditions for embryogenic calli bombardment, selection, and regeneration here optimized for sugarcane cv. C1051-73 were combined in a trans formation procedure that follows essentially the steps described by Frank and Birch [14]. The established procedure was effectively used to generate transgenic plants without the occurrence of escapes or mosaicism. The acquired resistance to geneticin remained stable in soil-grown plants, either in the primary stalk or the sprouts, after two years of vegetative propagation (see next epigraph).
Levansucrase activity but no levan accumulation in transgenic sugarcane plants carrying the Gluconacetobacter diazotrophicus levansucrase gene (lsdA)
After having established an efficient procedure for the genetic transformation of sugarcane cv. C1021-73, we proceeded to investigate the feasibility of producing the polyfructan levan in transgenic plants. To this aim, the vacuolar targeting pre-pro-peptide of onion sucrose:sucrose 1-fructosyltransferase (1-SST) was fused to the mature part of G. diazotrophicus levansucrase (LsdA) [5] under the transcriptional control of the maize ubiquitin-1 promoter and the tNos terminator (plasmid pKUBI-LsdA). The functionally of the chimaeric expression cassette was first demonstrated in transient transformation experiments. Embryogenic sugarcane calli was bombarded with plasmid pKUBI-LsdA and evaluated for levansucrase activity after 48 h of gene delivery. The incubation of cell extracts with the substrate sucrose resulted in the synthesis of fructan of DP higher than10 and FOS, as determined by TLC analysis (not shown).
In stable transformation experiments, only two percent of the overall number (above 1000) of the calli bombarded with pKUBI-LsdA succeeded to grow smoothly when cultured in the dark during one month on Medium H supplemented with 2,4 D and geneticin 20 mg/L. The non-treated calli failed to multiply and completely necrotized after incubation on the selection medium. The transgenic calli regenerated into normal plantlets after subculturing on Medium H with geneticin 25 mg/L in the light. Southern blot analysis demonstrated the integration of the target gene (lsdA) in the genome of adult plants from thirteen independent clones grown in the absence of the selection antibiotic (Figure 2A). All the evaluated plants, except the non-transformed control, showed the hybridization band of 583 bp corresponding to the internal EcoRI fragment of lsdA. The persistence of the selection gene (npt II) in sprouts of field-grown sugarcane lines was confirmed by PCR analysis (Figure 2B). The twenty tested lines remained to carry both transgenes after two complete cycles of vegetative propagation, indicating that no mosaic plants were recovered after transformation.
We failed to detect in vivo production of fructans in leaves and stems of transgenic plants grown under field conditions until the harvesting period (Figure 3A). Remarkably, the reaction of the leaf extracts from clone 4 on sucrose (30%, w/v) resulted in levan formation but there was no evident accumulation of the β-2,1-linked FOS, as determined by TLC analysis (Figure 3B). This result confirms the expression of active LsdA in at least one transgenic plant. We cannot exclude the existence of levansucrase activity in the other transgenic clones, although undetectable in our experimental conditions. The strong endogenous invertase activity in the sugarcane leaf extracts could have caused the hydrolysis of not only the substrate sucrose but also the potentially synthesized FOS, preventing the in vitro formation of levan in lines expressing low lsdA levels.
The sucrose-rich vacuole that occupies the majority of the volume of the sugarcane stem parenchyma is the most adequate cell compartment for transgenic levan synthesis and accumulation. In this compartment the synthesized polymer would remain sequestered, minimizing its potential disruptive effects. By using the maize Ubi-1 promoter, Wu and Birch [23] succeeded to express a bacterial sucrose isomerase tailored for vacuolar compartmentation in transgenic sugarcane at levels sufficient to permit the in vivo synthesis of isomaltulose in mature leaves and stems.
In our experiments, we used the same promoter but a distinct vacuolar targeting signal for lsdA expression in sugarcane. The low expression level of LsdA in the transgenic lines impeded to evaluate whether the pre-pro-peptide of onion 1SST targeted the recombinant enzyme to the sugarcane vacuoles. In transgenic tobacco, the constitutive expression of the chimaeric 1sst-lsdA gene did allow high levan accumulation in mature leaves and stem, suggesting the proper localization of LsdA in the cell vacuole [5].
There are no reports of levan production in transgenic sugarcane. Levan accumulation has been reported in the cell vacuole of other transgenic crops even without in vitro detection of the levansucrase activity [24]. In our experiments we tested levan formation in 20 adult transgenic plants corresponding to at least 13 independent transformation events, which is the number of bombarded calli that regenerated geneticin-resistant plantlets. Only one clone showed evident LsdA activity, while none of them accumulated levan.
Gene silencing, low lsdA expression, non-vacuolar localization of the recombinant enzyme, substrate competition with endogenous invertases, and even the putative presence of inducible fructan 6-exohydrolases (6-FEH) activity are possible explanations for the lack of levan accumulation in the transgenic sugarcane plants. Surprisingly, a functional 6-FEH gene was identified in sugar beet, a sucrose rich, non-fructan producer plant [25]. Despite this finding, other authors reported the accumulation of levan to low levels (maximum 0.5% of dry weight) in leaves, storage roots and fibrous roots of transgenic sugar beet constitutively expressing the Bacillus subtilis levansucrase fused to the CPY yeast vacuolar targeting sequence [26].
The generation of new lines transformed with the here described construct pKUBI-LsdA, the use of other promoters and vacuolar-driving signals, as well as the modification of the tissue culture conditions aiming to avoid potential counterselection of high lsdA-expressing transformants during early steps of calli multiplication, may conduct to the achievement of levan production in transgenic sugarcane.
REFERENCES
1. Rae AL, Grof CPL, Casu RE, Bonnett GD. Sucrose accumulation in the sugarcane stem: pathways and control points for transport and compartmentation. Field Crop Res. 2005;92(2-3):159-68.
2. Hernández L, Arrieta J, Menéndez C, Vázquez R, Coego A, Suárez V, et al. Isolation and enzymic properties of levansucrase secreted by Acetobacter diazotrophicus SRT4, a bacterium associated with sugar cane. Biochem J. 1995;309(Pt 1):113-8.
3. Trujillo LE, Arrieta JG, Dafhnis F, Garcia J, Valdes J, Tambara Y, et al. Fructo-oligosaccharides production by the Gluconacetobacter diazotrophicus levansucrase expressed in the methylotrophic yeast Pichia pastoris. Enzyme Microb Technol. 2001;28(2-3):139-44.
4. Trujillo LE, Gómez R, Banguela A, Soto M, Arrieta JG, Hernández L. Catalytic properties of N–glycosylated Gluconacetobacter diazotrophicus levansucrase produced in yeast. Electr J Biotechnol. 2004;7(2):115-23.
5. Banguela A, Arrieta JG, Rodríguez R, Trujillo LE, Menéndez C, Hernández L. High levan accumulation in transgenic tobacco plants expressing the Gluconacetobacter diazotrophicus levansucrase gene. J Biotechnol. 2011;154(1):93-8.
6. Snyman SJ, Watt MP, Huckett BI, Botha FC. Direct somatic embryogenesis for rapid, cost–effective production of transgenic sugarcane (Saccharum spp. hybrids). Proc S Afr Sug Technol Ass. 2000;74:186-7.
7. Arencibia AD, Carmona ER, Tellez P, Chan M, Yu S, Trujillo LE, et al. An efficient protocol for sugarcane (Saccharum spp. L.) transformation mediated by Agrobacterium tumefaciens. Transgenic Res. 1998;7(3):213-22.
8. Enríquez GA, Vázquez-Padrón RI, Prieto-Samsónov DL, de la Riva G, Selman–Housein G. Herbicide-resistant sugarcane (Saccharum officinarum L.) plants by Agrobacterium–mediated transformation. Planta. 1998;206(1):20-7.
9. Santosa DA, Hendroko R, Farouk A, Greiner R. A rapid and highly efficient method for transformation of sugarcane callus. Mol Biotechnol. 2004;28(2):113-9.
10. Birch RG. Plant Transformation: Problems and strategies for practical application. Annu Rev Plant Physiol Plant Mol Biol. 1997;48:297-326.
11. Christensen AH, Quail PH. Ubiquitin promoter-based vectors for high-level expression of selectable and/or screenable marker genes in monocotyledonous plants. Transgenic Res. 1996;5(3):213-8.
12. Murashige T, Skoog F. A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plantarum. 1962;15(3):473-97.
13. Finer JJ, Vain P, Jones MW, McMullen MD. Development of the particle inflow gun for DNA delivery to plant cells. Plant Cell Rep. 1992;11:323-8.
14. Franks T, Birch RG. Development and optimization of microprojectile systems for plant genetic transformation. Aust J Plant Physiol. 1991;18(5):453-69.
15. Sambrook J, Fritsch EF, Maniatis T. Molecular cloning, a laboratory manual. New York: Cold Spring Harbor Laboratory Press; 1989. p.1659.
16. Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene. 1996;173(1 Spec No):33-8.
17. Doyle JJ, Doyle JL. Isolation of plant DNA from fresh tissue. Focus. 1990;12: 13-5.
18. Arrieta J, Hernandez L, Coego A, Suarez V, Balmori E, Menendez C, et al. Molecular characterization of the levansucrase gene from the endophytic sugarcane bacterium Acetobacter diazotrophicus SRT4. Microbiology. 1996;142 (Pt 5):1077-85.
19. Wise CS, Dimier RJ, Davis HA, Rist CE. Determination of easily hydrolyzable fructose units in dextran preparations. Anal Chem. 1955;27(1):33-6.
20. Elliott AR, Campbell JA, Brettell RIS, Grof CPL. Agrobacterium-mediated transformation of sugarcane using GFP as a screenable marker. Aust J Plant Physiol. 1998;25(6):739-43.
21. de Ruijter NCA, Verhees J, van Leeuwen W, van der Krol AR. Evaluation and Comparison of the GUS, LUC and GFP Reporter System for gene expression studies in plants. Plant Biol. 2003;5(2):103-15.
22. Raza G, Ali K, Mukhtar Z, Mansoor S, Arshad M, Asad S. The response of sugarcane (Saccharum officinarum L.) genotypes to callus induction, regeneration and different concentrations of the selective agent (geneticin-418). Afr J Biotechnol. 2010;9(51):8739-47.
23. Wu L, Birch RG. Doubled sugar content in sugarcane plants modified to produce a sucrose isomer. Plant Biotech J. 2007; 5(1):109-17.
24. Cairns AJ. Fructan biosynthesis in transgenic plants. J Exp Bot. 2003;54(382):549-67.
25. Van den Ende W, De Coninck B, Clerens S, Vergauwen R, Van Laere A. Unexpected presence of fructan 6-exohydrolases (6-FEHs) in non-fructan plants: characterization, cloning, mass mapping and functional analysis of a novel ‘cell-wall invertase-like’ specific 6-FEH from sugar beet (Beta vulgaris L.). Plant J. 2003;36(5):697-710.
26. Pilon-Smits EAH, Terry N, Sears T, van Dun K. Enhanced drought resistance in fructan-producing sugar beet. Plant Physiol Biochem. 1999;37(4):313-17.
Received in September, 2011.
Accepted for publication in November, 2011.
Lázaro Hernández. Centro de Ingeniería Genética y Biotecnología, CIGB. AP 6162, La Habana, Cuba. E-mail: lazaro.hernandez@cigb.edu.cu

{kind=link}