










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBiotecnología Aplicada
versión On-line ISSN 1027-2852
Biotecnol Apl vol.29 no.1 La Habana ene.-mar. 2012
RESEARCH
Sequence of the gene coding for the p26 protein from a Cuban strain of Equine Infectious Anemia Virus
Secuencia del gen que codifica para la proteína p26 de una cepa cubana del virus de anemia infecciosa equina
Mavys Díaz-Miranda1, Dania Vázquez-Blomquist2, Luis D Cruz2, Claudia Vasallo2, Tania Campos1, Juan E Pérez1, Dario Paneque1, Carlos A Duarte2
1Empresa Productora de Vacunas Virales y Bacterianas, Grupo Empresarial LABIOFAM. Ave. Independencia, Km 16½, Boyeros, La Habana, Cuba.
2Centro de Ingeniería Genética y Biotecnología, CIGB. Ave. 31 e/ 158 y 190, AP 6162, Cubanacán, La Habana, Cuba.
ABSTRACT
Equine Infectious Anemia Virus (EIAV) is the causal agent of equine infectious anemia. Plasma virus RNA levels and the fraction of infected cells are both very low during the chronic stage of EIAV infections, turning the amplification of viral genes into a technically demanding task. In this work, DNA was extracted from the spleen of a chronically infected animal previously immunosuppressed with prednisolone, amplifying by nested PCR and sequencing the segment of the gag gene that codes for the p26 protein. Phylogenetic analysis placed the Cuban strain closest to three Canadian strains. The translated protein sequence exhibits 7 differences with the most related Canadian strain (Can7) and 13 with the Wyoming strain. This is the first report of a partial nucleotide sequence from EIAV in Cuba and the Caribbean.
Keywords: Equine Infectious Anemia Virus, PCR, p26, sequence, spleen, Cuba, capsid.
RESUMEN
El virus de la anemia infecciosa equina (VAIE) es el agente causal de la anemia infecciosa equina. Los bajo niveles de ARN viral en plasma y la baja proporción de células infectadas en la etapa de infección crónica hacen extremadamente difícil amplificar los genes virales. En este estudio se extrajo ADN del bazo de un portador crónico del virus, previamente sometido a un tratamiento inmunosupresor con prednisolona. Se amplificó un fragmento del gen gag que codifica para la proteína p26, mediante reacción en cadena de la polimerasa anidada, y se determinó su secuencia nucleotídica. Los análisis filogenéticos mostraron que esta cepa posee homología con tres cepas de Canadá. Se encontraron 7 aminoácidos diferentes con respecto a la cepa canadiense más similar (Can7) y 13 en relación con la cepa Wyoming. Este es el primer informe de la secuencia nucleotídica parcial de una cepa del VAIE circulante en Cuba y en el Caribe.
Palabras clave: Virus de la Anemia Infecciosa Equina, RCP, p26, secuencia, bazo, Cuba, cápsida.
INTRODUCTION
Equine Infectious Anemia Virus (EIAV) is a member of the Lentivirus genus, in the Retroviridae family. Its genetic organization closely resembles that of type 1 Human Immunodefi ciency Virus (HIV-1) [1, 2]. EIAV is the causal agent of equine infectious anemia (EIA), a disease restricted to members of the Equidae family that is distributed worldwide, causing important economic losses [2, 3]. The EIAV virion contains two glycoproteins in its envelope and four non-glycosylated internal proteins forming its capsid. Protein p26 is the most abundant capsid protein, exhibiting the highest sequence conservation not only among EIAV gene products, but among the capsid proteins of all retroviruses [4, 5].
Acute EIAV infections are characterized by frequent cycles of fever and intense viral replication, whose intensity and frequency gradually taper off until the host becomes an asymptomatic carrier for the remainder of its life. The absence of clinical symptoms, undetectable levels of viral RNA in plasma and low frequencies of infected cells are all signs of the strong control exerted at this stage on viral replication by the host immune system, which restricts the virus to organs of the lymphopoietic system [2, 6-9]. It has been shown that the administration of immunosuppressive drugs to asymptomatic carriers leads to the appearance of clinical signs resembling an acute infection and to increased viral replication [9-11].
No prophylactic vaccines are available against this disease. Since control of EIA takes a heavy financial toll, consisting mainly in the detection and elimination of infected animals [12, 13], a number of countries prefer to isolate these horses in segregated facilities. Such a counter-epizootic measure requires, however, the availability of sensitive and reliable diagnostic means. Agar gel double radial immunodiffusion (AGID), also known as Coggins test, is the technique recommended for this purpose by the International Office of Epizootics.
The application of molecular biology techniques such as reverse transcription followed by the polymerase chain reaction (RT-PCR), the latter often performed as a nested PCR, has allowed detecting and quantifying viral RNA in the plasma of recently or persistently infected animals. Nested PCR designs have also been employed to detect proviral DNA in monocytes/macrophages [14-18]. These techniques are also useful for genotyping wild strains and following up experimental infections, and can replace serological tests in the presence of interfering colostral antibodies [5, 9, 17].
Little is known, however, about the molecular epidemiology of this retrovirus, as the low levels of viral replication in chronically infected animals pose a formidable challenge to viral RNA detection in plasma [19-20]. Sequence databases contain a measly total of 17 sequences obtained from independent strains, originating in just 6 countries.
In Cuba, this disease has been detected in the provinces of Pinar del Río, Sancti Spíritus, Ciego de Ávila, Camagüey, Las Tunas, Holguín and Granma. Some of them relocate positive horses to segregation farms as an epizootic countermeasure, and the authorities in infection-free provinces usually prefer to sacrifice those horses they suspect might be infected. EIA is traditionally diagnosed by AGID, and no sequence information is available on circulating strains.
The purpose of the present study was to begin the genetic characterization of EIAV strains circulating in Cuba. Genomic DNA (gDNA) was isolated for this purpose from peripheral blood mononuclear cells (PBMC) and splenocytes of EIAV-infected horses that were previously subjected to an immunosuppressive treatment. Then, nested PCR was used to isolate a fragment of the gag gene coding for the p26 protein that was then sequenced. This constitutes the first report of the sequence of a circulating EIAV strain for Cuba and the Caribbean basin.
MATERIALS AND METHODS
Horses and clinical parameters
Four horses naturally infected with EIAV were included in this study. They were all asymptomatic carriers from a segregation farm in the Pinar del Río province, fi rst diagnosed with equine infectious anemia (EIA) by AGID, and declared negative for Leptospira, Brucella and blood parasites. These horses were subjected to a transitory immunosuppressive treatment intended to activate viral replication. Morning and afternoon rectal temperature readings were taken one week before, and during the immunosuppressive treatment.
Immunosuppression
Two of the animals were administered daily intramuscular doses of 2 mg of prednisolone per kilogram of bodyweight, during five days. The other two animals received intravenous doses of 3.45 mg of prednisolone per kilogram of bodyweight, administered every other day. Blood samples were drawn immediately after the appearance of symptoms of the disease (fever, lethargy, anorexia and others, but mainly increased body temperature). The animal with the highest body temperature was euthanized at the end of the study.
Sample extraction and processing
A total of 10 mL of venous peripheral blood were drawn from the jugular vein, using the Vacutainer system with EDTA as anticoagulant (Becton Dickinson, USA). All horses had anti EIAV antibody titers above 1:16 by AGID.
Erythrocytes were lysed by osmotic shock with a lysis solution composed of 0.03 M ammonium chloride, 0.002 M KHCO3, 0.02 mM EDTA. The resulting peripheral blood mononuclear cells (PBMC) were washed twice with PBS before proceeding to the extraction of gDNA.
DNA purification
Genomic DNA (gDNA) was extracted from peripheral blood mononuclear cells (PBMC) and splenocytes from one chronically infected individual and another undergoing immunosuppression, using Tri-Reagent (Sigma, USA) and following instructions from the manufacturer. One milliliter of Tri-Reagent was used for each sample of five million PBMC. Spleen tissue samples (50 mg) were processed in a Tissue Lyser II (Qiagen, USA, manufactured by Retsch), also using 1 mL of Tri-Reagent per sample. Absorbance at 260 nm and the 260/280 absorbance ratio were determined with a NanoDrop 1000 spectrophotometer (Thermo Scientific Nano Drop Technologies LLC, USA), and the purified nucleic acids were visualized by electrophoresis in 1% agarose gels.
Polymerase Chain Reaction
Amplification by PCR of the pcna reference gene
PCR amplifi cation reactions were set up for the pcna (proliferating cells nuclear antigen) housekeeping gene in every analyzed sample, following the protocol described by Schiller et al. and the oligonucleotide primers described therein. These primers generate a 242 bp amplifi cation product. The target gene is highly conserved across all mammalian species [21].
Amplification by nested PCR of a fragment of the gag gene
The first reaction employed the oligonucleotides described by Langemeier et al. [17], which yield an 853 bp-long amplification product. Published amplification conditions were modified to implement a touch-down PCR as follows: an initial denaturation at 94 °C for 10 min, followed by ten cycles of denaturation at 94 °C (30 s), annealing at 60 to 51 °C, decreasing temperature by 1 °C per cycle (30 s) and extension at 72 °C (1 min), followed in turn by 25 cycles of denaturation at 94 °C (30 s), annealing at 50 °C (30 s) and extension at 72 °C (1 min). An extension step of 72 °C for 7 min was added at the end of the reaction.
The primers described by Rosatti et al. [22] were modifi ed for the second PCR reaction, which yields a 705 bp-long amplification product. Their sequences were: sense primer- 5’-CCAATCATGATAGATGGGGCTGGAAACAG-3’; antisense primer: 5’-AAGTGCTTTTGCCAATAACATCATCTTTTG-3’.
Conditions for the second PCR included an initial denaturation at 94 °C for 10 min, followed by 30 cycles of denaturation at 95 °C (30 s), annealing at 54 °C (30 s) and extension at 72 °C (30 s). An extension step of 72 °C for 7 min was added at the end of the reaction.
All PCR reactions were performed in a total volume of 25 μL, including 1 μL of template DNA containing approximately 500 ng, 12.5 μL of PCR Master Mix 2X, (Promega, USA), and 25 pmol of each oligonucleotide. Amplified products were visualized by electrophoresis in 0.8% agarose gels at 120 V for 30 min. in the presence of ethidium bromide (0.5 μg/mL) and using Bromophenol Blue as running dye. Electrophoretic runs always included a DNA ladder (Promega, USA) as molecular weight marker, negative controls from the PCR, reagent controls and a positive control, prepared with leukocyte DNA from peripheral blood of an animal with an experimental infection of the Wyoming (Wy) reference strain.
DNA sequencing
The amplifi ed product was diluted to a concentration of 50 ng/μL and sent to Macrogen (South Korea) for further purifi cation and sequencing, using the same primers utilized for PCR, diluted at 5 pmol/μL.
Phylogenetic analysis
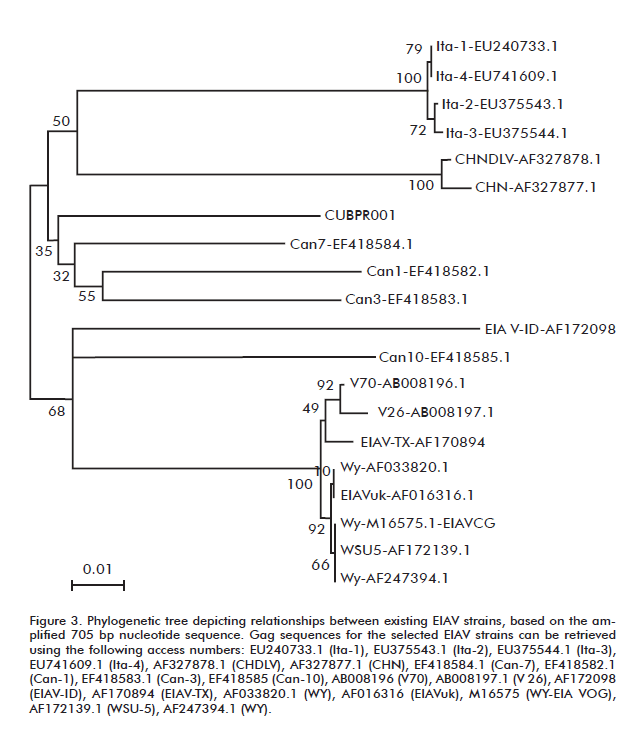
Nucleotide sequences were aligned using Mega 3.1 [23]. This software application is based on the Clustal X algorithm, which uses neighbor-joining [24] with Kimura’s two-parameter nucleotide substitution algorithm. The 705 nucleotide sequence of the amplified gag fragment was compared with existing EIAV GenBank sequences corresponding to this part of the gene. Their accession numbers, strain code and origin are: EU240733.1 (Ita-1), EU375543.1 (Ita-2), EU375544.1 (Ita-3) and EU741609.1 (Ita-4), all from Italy; AF327878.1 (CHDLV) and AF327877.1 (CHN) from China; EF418584.1 (Can-7), EF418582.1 (Can-1), EF418583.1 (Can-3) and EF418585 (Can-10) from Canada; AB008196 (V70) and AB008197.1 (V26) from Japan; and AF172098 (EIAV-ID), AF170894 (EIAV-TX), AF033820.1 (WY), AF016316 (EIAVuk), M16575 (WY-EIA VOG), AF172139.1 (WSU-5) and AF247394.1 (WY) from the United States.
RESULTS AND DISCUSSION
Although all four horses had clinical EIA symptoms (increased body temperature, anorexia and weakness), these were intense only in animal 5-04 (body temperature of 39.5 °C), indicating that higher doses of immunosuppressants or longer treatments must be employed in the future. Blood samples were drawn from all animals one day after concluding the treatment.
The samples were used to obtain PBMC, which were in turn used to extract genomic DNA. Template quality was assessed by PCR amplifi cation of the pcna gene from gDNA. A product of the expected size for this species (242 bp) [21] was observed in all cases, thereby ruling out poor sample quality as a factor accounting for failed EIAV amplifications.
These samples, however, failed to yield detectable amounts of the expected p26 amplicon upon EIAVspecific PCR, despite using the same conditions and oligonucleotides that successfully detect proviral copies in gDNA from PBMC obtained from horses experimentally infected with the Wyoming reference strain.
Other authors have had similar problems in the past [7, 19]. The amount of proviral DNA in PBMC from asymptomatic horses chronically infected with EIAV is known to be very low, bordering on the undetectable for all but the most sensitive of PCR setups [25-28]. We failed to obtain the p26 amplicon even in gDNA of PBMC from animals previously subjected to immunosuppressive treatments.
Harrold et al. [7] described differences in proviral DNA load for several organs (proviral DNA levels in spleen tissue, for instance, were 10- to 20-fold higher than anywhere else). Their results, in addition, confirmed that the levels of proviral DNA during chronic EIAV infections are extremely low, even in horses experimentally infected with the Wyoming strain that survive the acute stage of the disease.
It was decided, therefore, to try to amplify the proviral p26 sequence from gDNA extracted from the spleen of horse 5-04, which exhibited the most pronounced clinical symptoms. An amplicon with an electrophoretic mobility consistent with that of the pcna band was observed when this sample was amplified with the corresponding oligonucleotides (Figure 1). When subsequently amplified by means of a nested PCR with the same primer sets failing to produce a signal with PBMC gDNA, this sample yielded an amplicon whose electrophoretic mobility matched that of the expected 705 bp band (Figure 2).
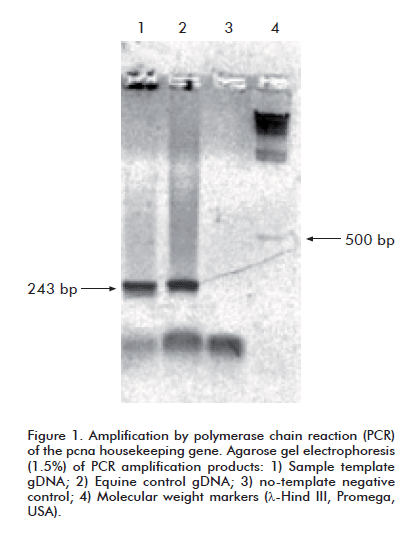
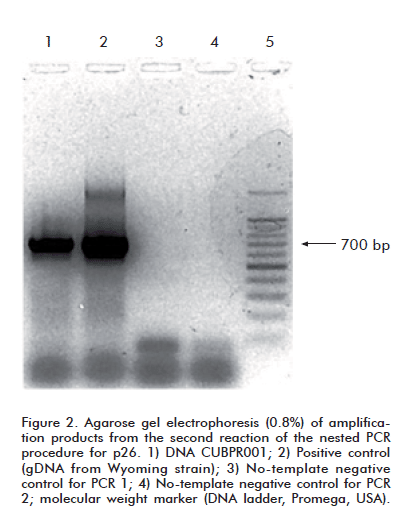
The amplified fragment was sequenced with the same primers used for the second PCR amplification. The obtained nucleotide sequence was denominated CUBPR001, and can be retrieved from GenBank under access number HQ853234.
Worldwide availability of EIAV sequence data is restricted to a total of only 17 independent strains, and most EIAV GenBank sequences correspond to viruses derived from the Wyoming reference strain. The only countries that report sequence information from indigenous EIAV strains corresponding to the gag segment amplifi ed in this work are USA, Canada, Argentina Italy, China, Japan and Thailand. A phylogenetic tree obtained by comparing the obtained 705 bp nucleotide sequence with that from these strains shows the genetic relations between the Cuban isolate and independent EIAV isolates from around the world (Figure 3).
It should be borne in mind that accuracy in phylogenetic studies increases with the number of sequences included in the analysis [29]. Our results are, therefore, constrained by the scarcity of available sequence data from independent strains.
However, phylogenetic tree reconstruction and estimation of evolutive parameters can also be improved by using longer sequences. In this sense, the 705 bp-long sequence used in the alignments is appropriate for obtaining reliable results when estimating genetic relatedness with distance-based methods, such as neighbor joining [24]. The analysis showed that strain CUBPR001 clustered closest to three of the ten Canadian strains, although bootstrap values in this node were low. Bootstrapping, used to estimate the level of confidence for internal branches of phylogenetic trees, is considerably simplified by the speed of the chosen analysis method [30].
Aiming at studying how the detected genetic differences translate into amino acid variability, it was decided to also prepare sequence alignments of translated p26 segments from the Cuban strain, the Wyoming strain, and the most closely related Canadian strain (Figure 4). There were 13 amino acid changes between the Cuban and Wyoming strains, and 7 between the Cuban and Can1 strains; confirming that the obtained p26 fragment is most similar, at the amino acid level, to viruses circulating in Canada.
The nested PCR setup used in this study has allowed obtaining, for the first time, sequence information from the gag gene of an indigenous Cuban EIAV strain. Combining PCR with a previous immunosuppressive treatment and extracting gDNA from spleen tissue seems to be the most effective procedure for the molecular characterization of new field isolates from chronically infected animals. Immunosuppression must, however, be further optimized, as increasing viremia to the point that amplification of viral sequences from PBMC becomes feasible would make invasive procedures, such as spleen biopsies, totally unnecessary, and would altogether obviate the occasional need to sacrifice the animal.
ACKNOWLEDGEMENTS
We would like to acknowledge the support of our colleagues at the Baró Basic Enterprise Unit, belonging to the Camilo Cienfuegos Genetic Enterprise, during sampling and set up of the immunosuppressive protocol. Our thanks also go to Dr. Santiago Dueñas and Dr. Liz Álvarez-Lajonchere, from the Hepatitis C department of the Center for Genetic Engineering and Biotechnology, for their help with the logistics of PCR set up and experimental execution, as well as fruitful technical advice.
REFERENCES
1. Li H, Zhang X, Fan X, Shen T, Tong X, Shen R, et al. A conservative domain shared by HIV gp120 and EIAV gp90: implications for HIV vaccine design. AIDS Res Hum Retroviruses. 2005;21(12):1057-9.
2. Maury W. Regulation of equine infectious anemia virus expression. J Biomed Sci. 1998;5(1):11-23.
3. Soutullo A, Santi MN, Perin JC, Beltramini LM, Borel IM, Frank R, et al. Systematic epitope analysis of the p26 EIAV core protein. J Mol Recognit. 2007;20(4):227-37.
4. Leroux C, Cadore JL, Montelaro RC. Equine Infectious Anemia Virus (EIAV): what has HIV’s country cousin got to tell us? Vet Res. 2004;35(4):485-512.
5. McConnico RS, Issel CJ, Cook SJ, Cook RF, Floyd C, Bisson H. Predictive methods to define infection with Equine Infectious Anemia Virus in foals out of reactor mares. J Equine Vet Sci. 2000;20(6):387-92.
6. Hammond SA, Li F, McKeon BM Sr, Cook SJ, Issel CJ, Montelaro RC. Immune responses and viral replication in longterm inapparent carrier ponies inoculated with equine infectious anemia virus. J Virol. 2000;74(13):5968-81.
7. Harrold SM, Cook SJ, Cook RF, Rushlow KE, Issel CJ, Montelaro RC. Tissue sites of persistent infection and active replication of equine infectious anemia virus during acute disease and asymptomatic infection in experimentally infected equids. J Virol. 2000;74(7):3112-21.
8. Leroux C, Craigo JK, Issel CJ, Montelaro RC. Equine infectious anemia virus genomic evolution in progressor and nonprogressor ponies. J Virol. 2001;75(10):4570-83.
9. Montelaro RC, Ball JM, Rushlow KE. Equine Retroviruses. In: Levy JA, editor. The retroviridae. New York: Plenum Press; 1993. p. 257-360.
10. Kono Y, Hirasawa K, Fukunaga Y, Taniguchi T. Recrudescence of equine infectious anemia by treatment with immunosuppressive drugs. Natl Inst Anim Health Q (Tokyo). 1976;16(1):8-15.
11. Tumas DB, Hines MT, Perryman LE, Davis WC, McGuire TC. Corticosteroid immunosuppression and monoclonal antibody-mediated CD5+ T lymphocyte depletion in normal and equine infectious anaemia virus-carrier horses. J Gen Virol. 1994;75 (Pt 5):959-68.
12. Nagarajan MM, Simard C. Gag genetic heterogeneity of equine infectious anemia virus (EIAV) in naturally infected horses in Canada. Virus Res. 2007;129(2):228-35.
13. Pare J, Simard C. Comparison of commercial enzyme-linked immunosorbent assays and agar gel immunodiffusion tests for the serodiagnosis of equine infectious anemia. Can J Vet Res. 2004;68(4):254-8.
14. Cook RF, Cook SJ, Li FL, Montelaro RC, Issel CJ. Development of a multiplex real-time reverse transcriptase-polymerase chain reaction for equine infectious anemia virus (EIAV). J Virol Methods. 2002;105(1):171-9.
15. Gendelman HE, Narayan O, Kennedy-Stoskopf S, Kennedy PG, Ghotbi Z, Clements JE, et al. Tropism of sheep lentiviruses for monocytes: susceptibility to infection and virus gene expression increase during maturation of monocytes to macrophages. J Virol. 1986;58(1):67-74.
16. Kim CH, Casey JW. Genomic variation and segregation of equine infectious anemia virus during acute infection. J Virol. 1992;66(6):3879-82.
17. Langemeier JL, Cook SJ, Cook RF, Rushlow KE, Montelaro RC, Issel CJ. Detection of equine infectious anemia viral RNA in plasma samples from recently infected and long-term inapparent carrier animals by PCR. J Clin Microbiol. 1996;34(6):1481-7.
18. Nagarajan MM, Simard C. Detection of horses infected naturally with equine infectious anemia virus by nested polymerase chain reaction. J Virol Methods. 2001; 94(1-2):97-109.
19. Oaks JL, McGuire TC, Ulibarri C, Crawford TB. Equine infectious anemia virus is found in tissue macrophages during subclinical infection. J Virol. 1998;72(9):7263-9.
20. Rice NR, Lequarre AS, Casey JW, Lahn S, Stephens RM, Edwards J. Viral DNA in horses infected with equine infectious anemia virus. J Virol. 1989;63(12):5194-200.
21. Schiller I, Huat LZ, Vaughan L, Weilenmann R, Koundrioukoff S, Pospischil A. Establishment of proliferative cell nuclear antigen gene as an internal reference gene for polymerase chain reaction of a wide range of archival and fresh mammalian tissues. J Vet Diagn Invest. 2003;15:585-8.
22. Rosati S, Profiti M, Lorenzetti R, Bandecchi P, Mannelli A, Ortoffi M, et al. Development of recombinant capsid antigen/transmembrane epitope fusion proteins for serological diagnosis of animal lentivirus infections. J Virol Methods. 2004;121(1):73-8.
23. Kumar S, Tamura K, Nei M. MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform. 2004;5(2):150-63.
24. Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406-25.
25. Kim CH, Casey JW. In vivo replicative status and envelope heterogeneity of equine infectious anemia virus in an inapparent carrier. J Virol. 1994;68(4):2777-80.
26. Maury W. Monocyte maturation controls expression of equine infectious anemia virus. J Virol. 1994;68(10):6270-9.
27. Sellon DC, Perry ST, Coggins L, Fuller FJ. Wild-type equine infectious anemia virus replicates in vivo predominantly in tissue macrophages, not in peripheral blood monocytes. J Virol. 1992;66(10):5906-13.
28. Sellon DC, Walker KM, Russell KE, Perry ST, Covington P, Fuller FJ. Equine infectious anemia virus replication is upregulated during differentiation of blood monocytes from acutely infected horses. J Virol. 1996;70(1):590-4.
29. Hillis DM. Inferring complex phylogenies. Nature. 1996;383(6596):130-1.
30. Page RDM, Holmes EC. Molecular evolution: a phylogenetic approach. Oxford: Blackwell; 1998.
Received in October, 2011.
Accepted for publication in January, 2012.
Carlos A Duarte. Centro de Ingeniería Genética y Biotecnología, CIGB. Ave. 31 e/ 158 y 190, AP 6162, Cubanacán, La Habana, Cuba. E-mail: carlos.duarte@cigb.edu.cu.


{kind=link}
{kind=link}