










My SciELO
 Custom services
Custom servicesServices on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.29 no.2 La Habana Apr.-June 2012
RESEARCH
Generation of two polyclonal antibodies for western blot detection of hepatitis A virus in plants
Generación de dos anticuerpos policlonales para la detección mediante western blot de virus de la hepatitis A en plantas
Abel Hernández1, Annabel González2, Alina López1, Yamilka Rosabal1, Yanaysi Ceballo1, Jeny Soto1, Gil Enríquez1
1 Bioreactors Laboratory, Plant Department, Center for Genetic Engineering and Biotechnology, CIGB PO Box 6162, Cubanacán, Havana, Cuba.
2 Virology Department, Biological Pharmaceutical Laboratories, LABIOFAM, Havana, Cuba.
ABSTRACT
In recombinant systems, the availability of good quality antibodies is required to detect Hepatitis A virus (HAV) proteins. Following this line, the cDNA sequences coding for two hepatitis A virus structural proteins, VP2 and VP3, were cloned into the bacterial expression vector pTrcHis yielding plasmids pTrcVP2 and pTrcVP3. Recombinant Escherichia coli XL1-blue strains harboring these two constructs were grown in liquid media and after IPTG gene induction, both fused recombinants molecules were detected by SDS-PAGE as 34 kDa and 24 kDa bands respectively. Recombinant VP2 and VP3 proteins containing N-terminal hexa-histidine tags were purified under denaturing conditions by immobilized metal affinity chromatography yielding 8 mg and 10 mg of refolded protein per 300 mL of bacterial culture respectively. Immunization of rabbits against purified recombinant proteins allowed the production of high titer polyclonal sera with immunological reactivity detecting hepatitis A virus proteins using western-blot analysis. According to our results, these two polyclonal sera constitute a valuable tool to follow the production of HAV proteins in transgenic plants, where a putative expression of HAV proteins higher than 0.25% of the total soluble protein could be detected with the use of these sera by western blot assay.
Keywords: Hepatitis A virus, structural protein, polyclonal sera.
RESUMEN
Para detectar las proteínas del virus de la hepatitis A en sistemas recombinantes se requiere de anticuerpos de calidad. Siguiendo estos objetivos la secuencia de ADNc que codifica para dos proteínas estructurales del virus de la hepatitis A, VP2 y VP3 se clonaron en el vector de expresión en Escherichia coli pTrc His resultando los plásmidos pTrcHisVP2 y pTrcHisVP3. Las células recombinantes portadoras de ambas construcciones fueron crecidas en medio líquido y después de la inducción con IPTG se detectaron ambas proteínas de 34 kDa y 24 kDa mediante SDS-PAGE. Las proteínas recombinantes VP2 y VP3 que contenían el tallo de histidina fueron purificadas en condiciones desnaturalizantes mediante cromatografía de afinidad a iones metálicos rindiendo 8 y 10 mg por 300 mL de cultivo respectivamente. La inmunización de conejos con las proteínas purificadas permitió la obtención de anticuerpos policlonales que permitieron la detección de las proteínas estructurales del virus de la hepatitis A en ensayos de western blot. De acuerdo a estos resultados los antisueros obtenidos pudieran ser útiles en el seguimiento en la producción de proteínas del virus de la hepatitis A en plantas transgénicas, donde una posible expresión de las proteínas del VHA superiores al 0.25% de las proteínas totales solubles podrían ser detectadas con estos sueros policlonales mediante ensayos de western blot.
Palabras clave: Virus de la hepatitis A, proteína de estructura, sueros policlonales.
INTRODUCTION
Hepatitis A virus (HAV) is a sole member of the family Picornaviridae and the causative agent of hepatitis A [1]. The HAV long open reading frame (ORF) encodes a polyprotein of approximately 250 kDa that undergoes co- and post-translational processing into smaller structural (VP4, VP2, VP3, and VP1-2A) and non-structural (2B, 2C, 3A, 3B, 3C, and 3D) proteins [2].
In spite of inactivated and attenuated vaccine against HAV are available, still there persist strategies that look for the recombinant antigen. The obtainment of HAV particles in recombinant system is a complex process because it requires processing large polyproteins and assembling subviral particles. Anti HAV antibodies are important and useful tools to characterize HAV antigen produced in a number of expression systems, including insect cells infected by recombinant baculoviruses [3], vaccinia viruses [4] and plant cells [5, 6].
In this study, the recombinant VP2 (rVP2) and VP3 (rVP3) proteins of HAV were expressed in Escherichia coli based on the cDNA sequences previously cloned in our laboratory [5]. They were purified by a zinc-charged chromatography column and used to produce rabbit polyclonal sera against both proteins. Here we report the production of a high-titer polyclonal sera specific against the rVP2 and rVP3, and its usefulness in the detection of HAV proteins in a western blot assay. The potential use of these sera to characterize HAV expression in plants was shown through immunoblotting analysis of tobacco extracts containing HAV particles.
MATERIALS AND METHODS
Plasmid constructs
The coding region of structural proteins VP2, VP3, VP1, 2A and nonstructural region 3ABC of HAV polyprotein were previously cloned yielding plasmid pΔMALm [5]. From this construct, two additional DNA plasmids were generated using the expression vector pTrcHisC (Invitrogen, USA), a pBR322-derived vector designed for efficient recombinant protein expression and purification in E. coli. Standard cloning procedures were followed: Plasmid pTrcVP2 was generated by cleaving pΔMALm from the Sma I site in the pBScript backbone vector to the Sca I site in the HAV genome. The resulting 700 bp fragment containing the complete ORF of the VP2 protein and the coding regions of the first nine aminoacids of VP3 protein was blunt-end ligated into pTrcHisC vector previously digested with BamH I and further blunt-ended. A second construct named pTrcVP3 was created by cloning the 575 bp fragment from plasmid pΔMALm containing 75% of the VP3 protein coding regions from the Sca I to the Hind III site of the ORF into the BamH I blunt-ended and Hind III sites of pTrcHisC vector. Competent E. coli XL1-blue cells were transformed with the constructs described above and correctly oriented clones were selected by restriction analysis with adequate enzymes.
Expression of rVP2 and rVP3 proteins in E. coli
Single colonies of E. coli strain XL1-blue containing the expression plasmid pTrcVP2 and pTrcVP3 were used to inoculate 50 mL of the Luria-Bertani (LB) medium containing 100 μg/mL ampicillin and 1% glucose. After 16 h of growth at 37 ºC, 20 mL, samples were inoculated into fresh 300 mL LB containing 100 μg/mL ampicillin. The bacterial cultures were incubated in a rotating orbital shaker at 37 ºC until reaching a 0.5 to 0.6 optical density at 600 nm. Expression of rVP2 and rVP3 was induced by addition of isopropyl-β,D-thiogalactopyranoside (IPTG; Applichem, Germany) at a final concentration of 1 mM. The bacterial cultures were incubated for an additional 5 h to allow suitable protein expression levels. Then, cultures were harvested by centrifugation at 1500 x g for 15 min at 4 ºC.
Purification of recombinant proteins
Thirty grams of bacterial pellets were resuspended in 30 mL 10 mM Tris, pH 8.0 (lysis buffer) and the cells were disrupted in French Press (Othake, Japan) at 1500 kgf/cm2, with two passes at 4 ºC. The cell lysates were separated into the soluble and insoluble fractions by centrifugation at 15 000 x g for 20 min at 4 ºC, and 10 µL were analyzed by SDS-PAGE under denaturing conditions. Both insoluble fractions resulting from rVP2 and rVP3 cell extracts were exposed to similar processes. Pellets were washed twice with 30 mL lysis buffer. Pellets were solubilized in 30 mL of 6 M Urea, made up in buffer lysis. The extraction was performed by gentle agitation during 1 h at 4 ºC and the supernatants were collected after centrifugation at 15 000 x g for 20 min at 4 ºC.
Subsequent purification of the rVP2 and rVP3 were performed on an immobilized metal affinity column (IMAC) using Chelating Sepharose Fast Flow resin (Amersham Pharmacia, Sweden) under denaturing conditions in similar form. It was loaded with 100 mM ZnSO4, washed with 10 mM Tris, 200 mM NaCl, 6 M Urea and 250 mM Imidazole, pH 8.0 (elution buffer), to elute weakly bound metal ions, also it was equilibrated with 10 mM Tris, 200 mM NaCl, 6 M Urea, pH 8.0 (equilibration buffer). Recombinant proteins solubilized in urea were applied to the IMAC column in both cases. The column was washed with 10 mM Tris, 200 mM NaCl, 6 M Urea and 50 mM Imidazole, pH 8.0, to eliminate contaminants. Recombinant proteins were eluted using the elution buffer. For all IMAC runs, the amount of protein loaded per volume of gel was 1 mg/mL. The yield of the recombinant protein was quantified using a BCA protein assay kit (Biorad, USA), using bovine serum albumin as standard. Proteins were renatured by sequential dialysis against decrescent urea concentration in phosphate saline buffer (PBS).
Production of sera against rVP2 and rVP3
Both recombinant proteins were used to produce polyclonal sera by repeated immunization of two female New Zealand White rabbits (CENPALAB, Cuba) around 3 kg, for each of the proteins tested. Three injections were performed on days 0, 21, 42 before the final bled on day 63. The first injection consisted of 0.5 mL of recombinant proteins (500 μg) in saline emulsified with 0.5 mL of complete Freund´s adjuvant (Sigma, USA). Subsequent injections used 200 μg of recombinant protein (0.5 mL) emulsified with 0.5 mL of incomplete Freund´s adjuvant (Sigma, USA). Injections were performed at multiple subcutaneous sites on the animals. All rabbits were maintained according to the Guide for the Care and Use of Laboratory Animals [7].
Immunoenzymatic assay
The recombinant proteins were diluted with a carbonate-bicarbonate buffer (15 mM Na2CO3, 30 mM NaHCO3, pH 9.6) containing 0.02% sodium azide, to a final concentration of 10 ng/mL. The plate was recovered with 1 ng of recombinant proteins during 1 h at 37 ºC. Subsequently, wells were incubated with rabbit sera (dilution in the range from 1/1000 to 1/640 000) followed by goat anti-rabbit IgG alka-line phosphatase conjugate (Sigma, USA) diluted 1/10 000. The reaction was revealed with the addition of each well of 100 μL of 4-Nitrophenyl disodium orthophosphate (BDH, England) prepared at a concentration of 1 mg/mL. The plate was incubated for 15 min and the reaction was stopped with 100 mM EDTA pH 8.0. The optical density was determined at 405 nm in a microtiter plate reader (Tecnosuma, Cuba).
The competition assay was carried out with a commercial kit (Mediagnost, Alemania) according to the manufacturer instructions and containing the standard sera, dilution buffer, diluted conjugate, substrate, washing and stop solutions. Briefly, sample, positive and negative standard sera were diluted 1/10 with dilution buffer. One hundred microliters were added to the wells of a microtiter plate, which have been previously coated with inactivated HAV antigen, and incubated for 2 hours at 37 °C. Subsequently, 50 μL of the diluted conjugate (peroxidase labeled anti-HAV IgG) were added and plates incubated again for 1 h at 37 °C. The excess conjugate was washed off and 100 µL of the substrate solution were added. Then, plates were incubated for 30 min at room temperature. The reaction was terminated by adding the stop solution. The optical density (OD) of the colored reaction product was measured on a microtiter plate reader (Tecnosuma, Cuba) at 450 nm.
To determine the neutralizing nature of antibodies obtained in rabbits against both structural proteins, neutralization limit (NL) was determined as follows:
![]()
Sera with OD values above and below the NL were considered as lacking neutralization capacity and neutralizing, respectively. Preimmune serum from rabbit 2 was used as negative control and the positive control was taken from the kit.
SDS-PAGE and Western blot analysis
To confirm the rVP2 and rVP3 expression, 100 µL of cell culture after 5 h post induction with IPTG were centrifuged a 5 000 x g. The pellet was resolubized and boiled for 30 min, separated in 12.5% SDS-PAGE followed by a Coomassie staining and a transfer to PVDF membrane (Whatman, USA). Blots were blocked in 5% skim milk in PBS before incubation with a dilution 1/10 000 of horseradish peroxidase labeled monoclonal anti-His tag (Sigma, USA). Membrane incubations were performed at 37 ºC during 1 h. Detection of recombinant protein was confirmed using diaminobenzidine (Sigma, USA) as substrate. Expression levels were determined by gel densitometry from a Coomassie-stained acrylamide gel.
One microgram of purified recombinant protein and 20 µL (200 ng) of HAVAC vaccine (Shenzhen Mellow Hope, China), were separated in 12.5% SDS-PAGE and transferred onto PVDF membrane. Detection of proteins was performed with sera against recombinant proteins diluted 1/200 in PBS and subsequent incubation of blots with goat anti-rabbit IgG-alkaline phosphatase conjugate diluted 1/10 000 (Sigma, USA). Membrane incubations were performed at 37 ºC for 1 h. The reaction was revealed with NBT/BCIP solution (Promega, USA) as substrate for this enzyme.
Functional assay and detection limits of the generated antibodies in Western blot assays
Nicotiana tabacum leaves were ground with mortar and mixed 1/1 (w/v) with PBS supplied with 0.1% Tween 20 (Merck, Germany). The total soluble proteins (TSP) extracted from tobacco leaves were mixed with concentrations of HAV proteins ranging from 0.03 to 2% of expression of TSP. The resulting mixture was separated in 12.5% SDS-PAGE. After the electrophoretic separation, proteins were transferred to a PVDF membrane. Detection of HAV proteins with sera against rVP2 and rVP3 was performed according to the description above.
RESULTS
Cloning and expression of VP2 and VP3 fragments
DNA fragments encoding VP2 and VP3 proteins were isolated from plasmid pΔMALm and cloned into the pTrcHisC vector for expression in E. coli as described in Materials and methods. DNA inserts are positioned in frame with a sequence that encodes an N-terminal fusion peptide that includes six histidine residues in series which function as a metal-binding domain in the translated protein. The resulting plasmids were named pTrcHisC-VP2 and pTrcHisC-VP3 (Figure 1A). Positive clones were selected by restriction analysis.
IPTG induction of E. coli transformed with empty pTrcHisC vector did not express the recombinant proteins while induced cultures of the recombinant strains resulted in the expression of two proteins of 34 kDa (rVP2) and 24 kDa (rVP3) respectively. Gel densitometry analysis revealed expression levels around 8% and 11% for both recombinant proteins (Figure 1B) with respect to TSP. An immunoblot a-nalysis was performed to confirm the identity of the proteins. The same bands previously described were detected by specific anti-hexahistidine tag antibody, although some lower bands were also evident which, we hypothesize, are degradation products (Figure 1C).
rVP2 and rVP3 extraction and purification
Fractionation of the bacterial lysates by high speed centrifugation into supernatant and pellet fractions revealed that the majority of both recombinant proteins were located in the pellet, presumably as inclusion bodies. Both proteins were capable to be soluble in the presence of 6 M urea. After purification through Chelating Sepharose Fast Flow resin, previously coupled with Zn2+ ion, rVP3 was obtained with 91% purity as estimated by gel densitometry analysis (Figure 2). Similarly, rVP2 was purified and its purity was estimated as 88% by the same method (data not shown). Since both purification procedures were under denaturing conditions, the proteins were renatured by sequential dialysis against lower urea concentrations, yielding 8 mg and 10 mg of refolded protein per 300 mL of culture respectively.
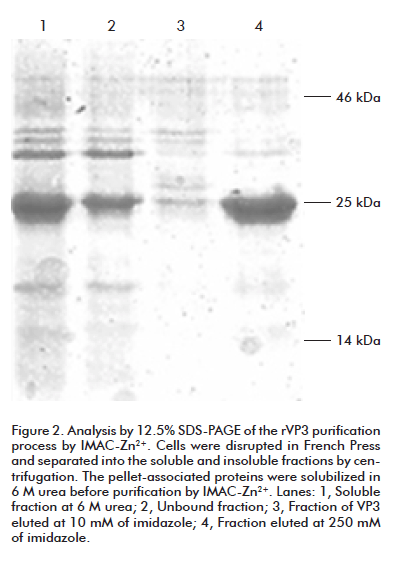
Rabbit immunization with rVP2 and rVP3
Recombinant proteins were used to immunize two rabbits for each case, by a three-dosage scheme. Polyclonal sera were evaluated by ELISA using 1 ng of recombinant protein to coat the microplate. Sera was serially diluted to determine the sera highest dilution which ELISA value was 3 times the value of preimmune serum. For both proteins, the highest serum dilution was 1/80 000 (Figure 3). For further analyses, rabbit serum 2 (against rVP2) and rabbit serum 4 (against rVP3) were selected.
A competition assay using a kit for the detection of antibodies against the HAV was carried out. This ELISA would detect antibodies able to recognize the neutralizing sites of the HAV and in this way, to block them prior to later incubation with antibodies whose neutralizing capacity is known. According to this, NL was determined as described in Materials and methods, based on the OD values shown in figure 4, for a final value of 1.75.

According these results, we can conclude that these sera do not recognize neutralizing epitopes of HAV.
We evaluated the recognition capacity of immune sera to HAV structural proteins by western blotting (Figure 5). The Chinese vaccine HAVAC, prepared from an attenuated viral formulation, was used as positive control. The estimated molecular weights for rVP2 and rVP3 protein are 24.8 and 27.8 kDa respectively, according to their amino acid sequence. Immune serum raised against rVP2 recognized a protein of 29 kDa in a viral sample, a slightly higher size compared to the theoretical estimated size (Figure 5B, line 2). This serum recognized the rVP2 protein at a size of 34 kDa according the expected size and showed slight recognition of rVP3 applied in a same lane (Figure 5B, lane 1).
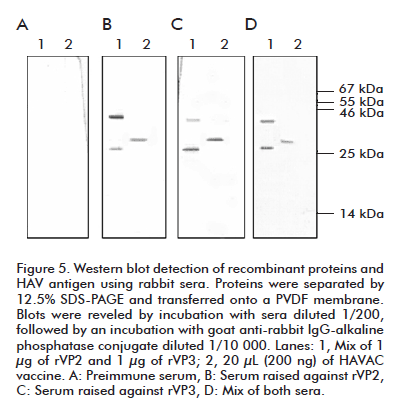
The serum raised against the rVP3 protein recognized in a viral preparation a protein of similar size to that observed during the incubation with rVP2 serum (Figure 5C, lane 2) and with the expected size. Recombinant VP3 polyclonal serum also slightly recognized rVP2, in a similar way to that described previously for the serum raised against rVP2 (Figure 5C, line 1). When incubating the PVDF membrane with a mix of both sera, only one band of 29 kDa was detected in the viral sample (Figure 5D, lane 2) while both recombinant proteins were observed in their expected size (Figure 5D, lane 2). No recognition of recombinant or viral proteins was observed when blots were incubated with preimmune sera (Figure 5A).
Functional assay and detection limits of the generated antibodies
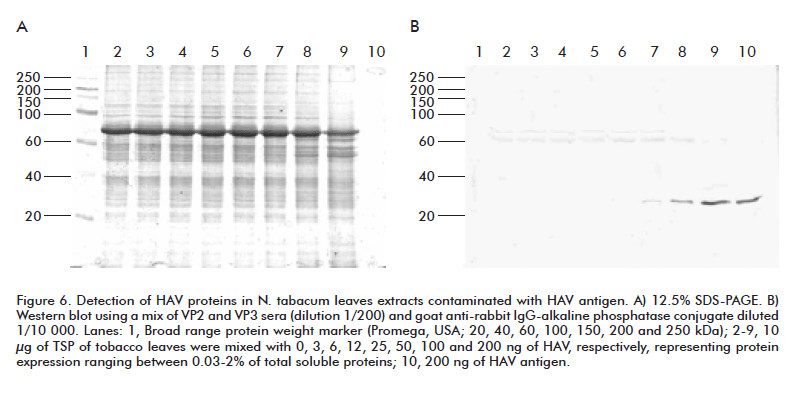
To evaluate the ability of the generated antibodies to recognize structural proteins in HAV particles, extracts from wild-type N. tabacum leaves were mixed with concentrations of HAV proteins ranging from 0.03 to 2% of expression of TSP. The resulting mixture was separated in 12% SDS- PAGE, and western blotting assay with rVP2 and rVP3 anti-sera to know the putative expression levels that could be detected by using these antibodies was done (Figure 6). Expression levels higher than 0.25% of TSP could be detected in the working dilutions used. Sensitivity could be increased given the lack of recognition of endogenous plant proteins with these antibodies at the region in which HAV proteins migrate in SDS-PAGE.
DISCUSSION
The main goal of this work was the obtainment of polyclonal antibodies (PAbs) that could be useful for analyzing HAV expression in plants. For this purpose, fragments of two structural proteins of the virus were cloned and expressed in E. coli. After purification, rabbit sera were produced against rVP2 and rVP3, and further evaluated under denaturing and non-denaturing conditions for specificity against the recombinant proteins and native HAV antigen. Despite of monoclonal antibodies against HAV have been produced and largely characterized [8, 9], polyclonal sera against HAV and structural proteins are suitable for laboratory use and epidemiological studies of HAV infection. PAbs can be generated by procedures faster, less expensive and technically simpler than required to produce monoclonal antibodies [10]. In general, the generation of PAbs is based on immunizing animals with proteins isolated from natural sources or produced by recombinant techniques. The latter route has been extensively used and PAbs obtained are common tools for diagnostics [11] or protein expression profile analysis [12]. Furthermore, the ability of polyclonal reagents to recognize a multiplicity of epitopes is significant in some immunological assays, where the detection of a molecule could be compromised by recognizing a single epitope. This is especially important in transgenic plants, where the accumulation of heterologous protein is fundamental. Besides, the production of rVP2 and rVP3 in E. coli would facilitate to obtain monoclonal antibodies by immunizing mice with these antigens, if necessary.
To express rVP2, a segment of 700 bp was selected from plasmid pΔMALm[5], coding for the complete amino acid sequence of the structural VP2 protein and the first 9 amino acids of the VP3 coding sequence. This nucleotide sequence generated a 304-amino acid polypeptide that included on its N-terminus 36 amino acids corresponding to the histidines tag, XPress epitope and the cleavage site for enterokinase. This fusion protein has a theoretical molecular weight of approximately 34 kDa, unlike the native VP2 protein.
In the case of rVP3, a 575-bp segment was selected coding for 75% of the native VP3 amino acid sequence, generating a 201-amino acids polypeptide with a theoretical molecular weight of 27 kDa and including the N-terminal sequences of the expression vector. This segment conserved the majority of lineal epitopes present in the native VP3. Both proteins used in our study were obtained in insoluble form, presumably at inclusion bodies, since the cytoplasmic high-level expression of these proteins in E. coli frequently generates inclusion bodies. Strategies to solubilize an inclusion body and refold the protein into its native structure have been recently reviewed [13]. Here, we resuspended the recombinant proteins in 6 M urea and further purified them by IMAC under denaturing conditions based on the fused N-terminal His tag, finally refolding them by sequential dialysis at decreasing urea concentrations. Both proteins were properly folded by this process, since they remained soluble at the final dialysis step. In spite of being time-consuming, this refolding method renders high yields [14, 15]. The 88% and 91% of purity for rVP2 and rVP3 were respectively enough to produce high-titer polyclonal sera in rabbits.
Sera specificity was demonstrated by western blotting analysis. A slight cross-reactivity between rVP2 and rVP3 was seen. Keeping in mind that VP2 and VP3 don’t share common sequences in the HAV, this slight cross-reactivity could be explained by the domain shared by both recombinant proteins on their N-terminus by the His tag, the XPress epitope and the marginal recognition of the 9 amino acids of VP3 at the C terminus of the rVP2 (not detrimental cross-recognition for the purpose of antigen detection).
Sera against rVP2 and rVP3 didn’t recognize the major conformational epitope of HAV in a competition immunoenzymatic assay using the neutralizing antibody 7E7. However, we observed in this assay a slight blockade of the neutralization epitope when incubating with sera against rVP2 and rVP3, presumably due to the interaction with lineal sites present in viral particles. A similar effect of poor neutralization capacity of sera obtained from the injection of HAV recombinant proteins has been previously demonstrated [16, 17], even for the virus major structural protein VP1. This is the main cause of the search for the expression of HAV subviral structures to be used as vaccine candidates.
Anti-rVP2 and rVP3 sera recognized the structural proteins of HAV in a western blot analysis. When we incubated membrane with an anti-serum against rVP2, a band was detected corresponding to 29 kDa in a lane of viral preparation, in spite of the 24.8 kDa theoretical molecular weight for VP2. Previous studies have associated this delay in the migration of VP2 in polyacrylamide gels under normal conditions with this protein possibly being the VP2 precursor, the VP0 protein, which proteolytic processing constitutes the final step of the hepatitis A virion maturation [18]. The rVP2 protein was detected at a size of 35 kDa, in agreement with the addition of 36 amino acids from the expression vector.
Serum raised after immunization with rVP3 detected a protein band at the expected relative size corresponding to the predicted molecular weight. VP3 was also detected when the membrane was incubated with this serum. The theoretical molecular weight of VP3 is 27.8 kDa, similar to the size at which the VP2 protein was detected in the viral lane. As mentioned above, both proteins do not share common sequences in the HAV; however, they have similar migration patterns under SDS-PAGE without adding urea. This was corroborated by incubating the viral lane with a mix of sera against both recombinant proteins, while rVP2 and rVP3 were clearly detected as different protein bands. Ross and Anderson resolved this co-migration of VP0 and VP3 by adding urea to SDS-PAGE [19].
The obtainment of HAV particles in recombinant systems is a reliable way to develop an effective vaccine against this virus. Plants are emerging as a promising system to express and manufacture a wide range of functionally active proteins of high value for the health industry [20]. High-level protein expression is essential to develop economically competitive plant-based processes for cultivation of transgenic varieties within confined fields and under controlled environmental and biosafety contained conditions. We have previously obtained HAV particles in tobacco cells yielding expression levels in the range of 0.0015% of TSP [5] and we are developing strategies to increase those expression levels. The sera obtained in this study will be useful in the screening of transgenic plants expressing HAV proteins by western blotting.
REFERENCES
1. Gust ID, Coulepis AG, Feinstone SM, Locarnini SA, Moritsugu Y, Najera R, et al. Taxonomic classification of hepatitis A virus. Intervirology. 1983;20(1):1-7.
2. Harmon SA, Updike W, Jia XY, Summers DF, Ehrenfeld E. Polyprotein processing in cis and in trans by hepatitis A virus 3C protease cloned and expressed in Escherichia coli. J Virol. 1992;66(9):5242-7.
3. Dzagurov GK, Kusov I, Gauss-Mueller V. Expression of hepatitis A virus procapsids in the insect cells infected by recombinant baculovirus. Vopr Virusol 2003;48(3): 36-40.
4. Yao GFLC, Jiming RLZ. Expression of Hepatitis A virus proteins by recombinant vaccinia virus [J]. Chinese J Virol. 1989;4. Available from: http://en.cnki.com.cn/Article_en/CJFDTOTAL-BDXB198904000.htm
5. López A, Rosabal Y, Hernández A, González B, Ríos J, Pérez M, et al. Expression of the Hepatitis A virus empty capsids in suspension cells and transgenic tobacco plants (Nicotiana tabacum L.). Biotecnol Apl 2008;25(1):42-6.
6. National Research Council. Guide for the Care and Use of Laboratory Animals. 8th Edition. Washington, DC: The National Academies Press; 2011.
7. Chung HY, Lee HH, Kim KI, Chung HY, Hwang-Bo J, Park JH, et al. Expression of a recombinant chimeric protein of hepatitis A virus VP1-Fc using a replicating vector based on Beet curly top virus in tobacco leaves and its immunogenicity in mice. Plant Cell Rep. 2011; 30(8):1513-21.
8. Dawson GJ, Decker RH, Norton DK, Bryce WH, Whittington RO, Tribby, II, et al. Monoclonal antibodies to hepatitis A virus. J Med Virol 1984;14(1):1-8.
9. Schofield DJ, Emerson SU, Purcell RH. Four chimpanzee monoclonal antibodies that neutralize hepatitis A virus. Drugs Fut 2003;28(2):137-42.
10. Lipman NS, Jackson LR, Trudel LJ, Weis-Garcia F. Monoclonal versus polyclonal antibodies: distinguishing characteristics, applications, and information resources. ILAR J. 2005;46(3):258-68.
11. Lu L, Cheng A, Wang M, Jiang J, Zhu D, Jia R, et al. Polyclonal antibody against the DPV UL46M protein can be a diagnostic candidate. Virol J 2010;7:83.
12. Grifantini R, Pagani M, Pierleoni A, Grandi A, Parri M, Campagnoli S, et al. A novel polyclonal antibody library for expression profiling of poorly characterized, membrane and secreted human proteins. J Proteomics. 2011;75(2):532-47.
13. Burgess RR. Refolding solubilized inclusion body proteins. Methods Enzymol. 2009;463:259-82.
14. Jang TH, Park HH. Generalized semi-refolding methods for purification of the functional death domain superfamily. J Biotechnol. 2011;151(4):335-42.
15. Zhang Y, Ma Y, Yang M, Min S, Yao J, Zhu L. Expression, purification, and refolding of a recombinant human bone morphogenetic protein 2 in vitro. Protein Expr Purif. 2011;75(2):155-60.
16. Johnston JM, Harmon SA, Binn LN, Richards OC, Ehrenfeld E, Summers DF. Antigenic and immunogenic properties of a hepatitis A virus capsid protein expressed in Escherichia coli. J Infect Dis. 1988; 157(6):1203-11.
17. Gauss-Muller V, Zhou MQ, von der Helm K, Deinhardt F. Recombinant proteins VP1 and VP3 of hepatitis A virus prime for neutralizing response. J Med Virol. 1990; 31(4):277-83.
18. Gauss-Muller V, Lottspeich F, Deinhardt F. Characterization of hepatitis A virus structural proteins. Virology 1986; 155(2):732-6.
19. Ross BC, Anderson DA. Characterization of hepatitis A virus capsid proteins with antisera raised to recombinant antigens. J Virol Methods. 1991;32(2-3):213-20.
20. Tiwari S, Verma PC, Singh PK, Tuli R. Plants as bioreactors for the production of vaccine antigens. Biotechnology Advances. 2009;27(4):449-67.
Received in June, 2011.
Accepted for publication in May, 2012.
Abel Hernández. Bioreactors Laboratory, Plant Department, Center for Genetic Engineering and Biotechnology, CIGB PO Box 6162, Cubanacán, Havana, Cuba. E-mail: abel.hernandez@cigb.edu.cu.


{kind=link}
{kind=link}
{kind=link}