










My SciELO
 Custom services
Custom servicesServices on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.29 no.3 La Habana Jul.-Sept. 2012
RESEARCH
Generation of a murine model of chronic progressive experimental autoimmune encephalomyelitis for molecular pharmacology studies in Multiple Sclerosis
Generación de un modelo murino de encefalomielitis autoinmune experimental crónico progresivo para estudios de farmacología molecular en esclerosis múltiple
Karelia Macías-Cosme1, Majel Cervantes-Llanos1, Javier Marín-Prida2, Viviana Falcón-Cama1, Eduardo Pentón-Arias1, Giselle Pentón-Rol1
1 Centro de Ingeniería Genética y Biotecnología, CIGB. Ave. 31 e/ 158 y 190, Cubanacán, Playa, CP 11600, La Habana, Cuba.
2 Centro de Estudios para las Investigaciones y Evaluaciones Biológicas, CEIEB, Instituto de Farmacia y Alimentos, Universidad de La Habana. Ave. 23, e/ 222 y 214, La Coronela, La Lisa, La Habana, Cuba.
ABSTRACT
Experimental autoimmune encephalomyelitis (EAE), when induced in syngenic C57BL/6 mice by using myelin oligodendrocyte glycoprotein (MOG), usually exhibits a chronic progressive pattern. This model mimics many of the symptoms and clinical signs typical of multiple sclerosis (MS) in humans. The present work describes specific adjustments to the experimental parameters described in the literature that were necessary when implementing this model under our conditions, demonstrating the presence of EAE in experimental animals by means of clinical evaluations, molecular assays and ultrastructural studies. The disorder was induced by active immunization with peptide MOG35-55 emulsified in Freund’s Incomplete Adjuvant supplemented with Mycobacterium tuberculosis, together with two additional administrations of Pertussis toxin. All immunized animals exhibited the typical clinical signs of EAE, with a severity that increased progressively from day 8 post-induction to the end of the evaluation, at day 28 post-induction. The maximum clinical score was 3.5, and the disorder was not reversible. Body weight loss was associated with clinical deterioration at the initial stages of the experiment. From a molecular perspective, it was shown that effector cytokines (mainly IL-17) were positively regulated in the brain of diseased animals. The characteristic ultrastructural changes of MS were also detected; namely, demyelination and axonal damage. The methodology described here enabled the implementation of an animal model that reproduces fundamental aspects of the pathogenesis of MS and is, therefore, highly useful for the study of physiopathological mechanisms, the identification of new pharmacological targets and the evaluation of specific biomolecules with therapeutic purposes.
Keywords: experimental autoimmune encephalomyelitis, myelin oligodendrocyte glycoprotein, demyelination, multiple sclerosis, C57BL/6.
RESUMEN
La inducción de la encefalomielitis autoinmune experimental (EAE) mediante la glicoproteína de la mielina del oligodendrocito (MOG) en ratones C57BL/6 isogénicos y homocigóticos, se caracteriza por un patrón crónico progresivo. Este modelo remeda un amplio espectro de síntomas y signos clínicos típicos de la esclerosis múltiple (EM) en humanos. Se describe el ajuste de las condiciones experimentales con respecto a las referidas en la literatura, y se demuestra la generación del modelo mediante evaluaciones clínicas, moleculares y ultraestructurales. La enfermedad se indujo por inmunización activa con el encefalitógeno constituido por el péptido MOG35-55 emulsificado en adyuvante incompleto de Freund, suplementado con Mycobacterium tuberculosis y dos administraciones adicionales de toxina pertussis. Los animales inmunizados mostraron signos clínicos de EAE, cuya severidad aumentó en forma progresiva y ascendente a partir del día 8 después de la inducción, con un puntaje clínico máximo de 3.5, sin reversión hasta el día 28, en que finalizó el periodo de evaluación. La disminución del peso corporal estuvo asociada con el inicio del deterioro clínico. Desde el punto de vista molecular, se identificó una regulación positiva de citocinas efectoras en el cerebro de los animales enfermos, principalmente de la IL-17. Se observaron daños ultraestructurales característicos de la EM en la mielina y el axón. La metodología permitió la generación de un modelo animal con elementos esenciales de la patogénesis de la EM y, por tanto, útil para el estudio de mecanismos fisiopatológicos, la identificación de nuevos blancos farmacológicos y la evaluación de biomoléculas con fines terapéuticos.
Palabras clave: encefalomielitis autoinmune experimental, glicoproteína de la mielina del oligodendrocito, desmielinización, esclerosis múltiple, C57BL/6.
INTRODUCTION
Multiple Sclerosis (MS), a chronic demyelinating and neurodegenerative inflammatory disorder affecting close to 2.5 million persons, constitutes the most important cause of neurological disabilities in young adults [1], especially in developed countries of the northern hemisphere. During the intervening period between successive crises, the disorder is usually managed with interferon beta (IFN-β), Copaxone, Natalizumab or Fingolimod (Gilenya®), a recently approved oral therapy [2]. These drugs, however, are expensive (in the order of $ 20 000 to $ 60 000 dollars per patient per year) [3] and their therapeutic effect is relatively modest, so that finding better alternatives for this disorder has remained a highly competitive research field.
Procuring preclinical efficacy data is a fundamental step in the development of new drugs. For every existing therapy against MS, this stage has been completed with the help of animal models of experimental autoimmune encephalitis (EAE) [4]. In addition, EAE models have been essential for research on the modulation of immunoregulatory circuits, a field that has provided important results for our understanding of the pathogenesis of MS.
From a molecular perspective, EAE is a disorder mediated mainly by CD4+ T cells [5]. Although CD4+ Th1 cells were considered during a long time as the sole players in this stage, it is now acknowledged that interleukin 17 (IL-17) and 22 (IL-22)-secreting Th17 cells [6] also have a fundamental role in the inflammatory process and other molecular events characteristic of the pathogenesis of EAE [7, 8]. The entry of autoreactive T cells into the central nervous system, together with other inflammatory mediators, is facilitated by an increase in vascular permeability at the blood-brain barrier, contributing to the demyelination and axonal loss typical of EAE and MS [5].
EAE is usually induced in susceptible animal species by immunization with myelin sheath antigens. This procedure is not equally successful with every animal species, however –especially when using myelin oligodendrocyte glycoprotein (MOG) for this purpose– due to variations in genetic background and allele polymorphisms of class II histocompatibility complex molecules and T-cell receptors [9]. In addition, environmental factors can be determinant in the development of EAE. Instances have been described, to cite one example, where experimental animals have become completely impervious to the induction of EAE depending, solely, on the particular facilities or animal handling procedures employed during the experiment [10]. Many hypotheses have been put forward to explain this behavior, including, among others, the presence or intestinal parasites (the so-called ‘hygiene hypothesis’) [11].
Based on these precedents, we decided to carefully document the experimental conditions used for obtaining a model of chronic progressive EAE in C57BL/6 mice, given that its implementation depends on very precise adjustments to these values. This paper therefore describes all adjustments and procedures necessary to guarantee the reproducibility and trouble-free implementation of this model by other researchers of the field. A number of clinical and molecular variables, together with ultrastructural analyses, were used to demonstrate the implementation of the model. Our EAE model is robust and provides an adequate predictive value for developing new drugs against MS.
MATERIALS AND METHODS
Animals
This work employed a total of 30 female mice of the C57BL/6 line (National Center for the Production of Laboratory Animals, CENPALAB, Cuba) 5 to 6 weeks old, with a body weight of 16 to 18 g at the moment of immunization. They were used after a laboratory adaptation period of one week. The animals were kept under conditions of controlled temperature (19 ± 2 °C) and relative humidity (55-65%), using a light-darkness period of 12 h. They were fed sterile concentrated rodent chow (5 g/animal) and water ad libitum.
The animals were distributed randomly into three groups of 10 individuals each: a negative control group (mice receiving neither immunizations nor pharmacological treatment); a placebo-treated EAE group (immunized animals treated only with daily subcutaneous (s.c.) injections of sterile phosphate buffer) and a hydrocortisone-treated EAE group (immunized animals treated with daily s.c. injections of 1 mg/kg hydrocortisone dissolved in sterile phosphate buffer, PBS). The daily s.c. injections of hydrocortisone or placebo were administered from the starting date of the experiment until day 12 after induction. The experimental protocol was submitted for review to the institutional reviewing committee for the use and handling of laboratory animals of the Center for Genetic Engineering and Biotechnology (CIGB, Cuba), according to current international standards [12].
Induction of experimental autoimmune encephalomyelitis
We used essentially the method described by Pluchino et al. [13] with some modifications. The mice were immunized using alternating subcutaneous inoculations into the left or right flank of the animals at the thoracic level, in days 0 and 7. Each inoculation delivered 200 µg of the inductor antigen (a peptide corresponding to residues 35-55 of the MOG antigen; MOG35-55: MEVGWYRSPFSRVVHLYRNGK) with a purity higher than 98% (CIGB, Cuba). The MOG35-55 peptide was emulsified in 200 µL of Freund's Incomplete Adjuvant (FIA; Sigma, USA), supplemented with Mycobacterium tuberculosis (MT, strain H37Ra; Difco, USA) at a final concentration of 8 mg/mL. The mice were also inoculated intravenously with a solution of pertussis toxin (PT; 200 ng in 100 µL of PBS) through the tail vein, immediately after administering the inductor encephalitogen, at day 0 and then at day 2.
Preparation of the antigen for inducing EAE
FIA plus MT
The contents of one ampoule of MT (100 mg) were transferred into a small mortar and the yellowish bacterial aggregates were ground into a whitish fine powder and then mixed under continuous agitation with 12.5 mL of FIA. The mixture was transferred to a 50 mL tube that was stored under slow, constant stirring at 4 °C. This mixture can be preserved stable for a maximum of 2 months at 2-8 ºC before use.
MOG35-55 peptide
The MOG peptide was dissolved at 2 mg/mL (200 µg/100 µL) in sterile PBS, pH 7.4, and then sterilized by filtration through a 0.2 µm pore size membrane (Corning, Germany).
MOG: FIA + MT emulsion
A volume of MOG peptide was gradually added to a volume of FIA + MT (v/v) at room temperature, and kept under constant medium-speed mechanical stirring for 5 to 10 min, in order to obtain a water/oil emulsion that would increase the immunogenicity of the preparation. The mixture was then transferred to a pair of 5 mL glass syringes connected to each other through a 16 G metal capillary, carefully avoiding the inclusion or formation of air bubbles inside the emulsion. After transferring the mixture from one syringe to the other for 5 to 8 cycles, the capillary was replaced by an 18 G metal capillary and the process was repeated. Proper formation of the emulsion was verified by placing one drop onto a Petri dish containing PBS. Once the emulsion was obtained, the syringes were wrapped in aluminum foil and stored at 4 °C overnight. It is recommended to prepare the emulsion at most 12 hours before use, in order to minimize the possibility of phase separation.
One-milliliter plastic syringes were used for the immunizations, loaded directly from the glass syringe by means of the metal capillary. Two-hundred microliters were injected into each animal.
Preparation of PT
PT (isolated from Bordetella pertussis, Sigma, USA) was prepared 24 h before the immunizations. Two-hundred nanograms of PT were dissolved in 100 µL of sterile PBS, pH 7.4, and stored at 4 °C until used. This storage period was necessary in order to prevent the death of the animals associated with the use of freshly prepared PT. Each inoculation delivered a total of 200 ng of PT per animal.
Model evaluation
Clinical monitoring of EAE
The clinical severity of the disorder in all three experimental groups was monitored by daily double-blind observations, performed always at the same hour, by a trained observer, starting from day 0 and until day 28 after induction. Care was taken to avoid stressing the animals during handling as much as possible, minimizing environmental factors such as noise, pain and heat. The following evaluation scale was used, prepared from previous results of other authors in this model [14, 15]: 0, No apparent clinical signs; 0.5, crooked stance or mildly irregular walking motions; 1, Mild loss of tail tone or mild paraparesis (weakness) of one posterior limb; 1.5, Partial loss of tone of the tail and mild paraparesis (weakness) of both posterior limbs; 2, Complete tail paralysis; 2.5, Complete tail paralysis with marked weakness of both hind limbs and occasional failure of hindquarters; 3, Partial paralysis (hemiparalysis) of hind limbs, the animal is forced to crawl; 3.5, Full paralysis of hind limbs without residual limb movement; 4, Full paralysis of hind limbs with weakness of anterior limbs; 4.5, Moribund and 5, Dead. An average clinical index was calculated for each group based on the individual scores, and the maximum clinical index for each animal during the study was also determined.
Measurement of body weight
Body weight for animals from the negative control group and the placebo-treated EAE group was evaluated daily, at the same hour every day, starting from day 7 and until day 28 after induction, using an analytical balance (Denver Instrument XP-3000, USA). The difference between the observed body weight and that measured at day 0 of the study was determined daily, calculating group means to follow the behavior of this variable.
Evaluation of effector and regulatory cytokines
At day 18 after induction, real time quantitative Polymerase Chain Reaction (qPCR) assays were run in two animals each from the negative control group and the placebo-treated EAE group, using three replicates per cytokine gene and β-actin as endogenous expression control. Messenger RNA was extracted from brain tissue as described by Lech et al. [16]. The reactions used SYBR Green Dye as detection system, and were run in a Light Cycler 480 (Roche, Mannheim, Germany) real time PCR cycler. All technical stages complied with the Minimal Information for Publication of qPCR Experiments (MIQE) guidelines [17]. Relative quantification was performed using the following formula [18]:
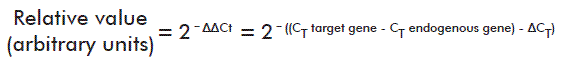
Where: CT represents the fractional cycle number at which the concentration of the target amplicon reaches a predetermined threshold; ∆CT represents the difference between the CT of target and reference genes (see [18] for more details).
Non-specific amplification was controlled by evaluating the target genes in comparison to the reference gene (β-actin). Specific oligonucleotide primers were employed at 300 nM (Metabion, Martinsried, Germany; see Table). Their design employed a step of in silico specificity screening with the BLAST search and alignment algorithm [19].
Ultrastructural study by transmission electron microscopy
At the end of the study period, three animals each were randomly selected from the negative control and placebo-treated EAE groups and sacrificed by cervical dislocation. The euthanized animals were dissected to take spinal cord samples that were fixed in glutaraldehyde or paraformaldehyde (1 and 4%, respectively, from Polysciences, USA) for 1 h at 4 °C, washed then in 0.1 M sodium cacodylate, pH 7.4 (BDH Chemicals, UK), and fixed in 1% osmium tetroxide (Agar Scientific, USA) for 1 h at 4 °C. The samples were then dehydrated by treatment with increasing concentrations (30, 50, 70 and 100%, 10 min at 4 °C for each) of ethanol (BDH Chemicals, UK) and included according to Spurr [20]. Ultrathin sections (40 to 50 nm) obtained with an ultramicrotome (NOVA, LKB, Germany) were placed on 400-mesh nickel grids (Agar Scientific, USA), stained with saturated uranyl acetate (Polysciences, USA) and lead citrate (Agar Scientific, USA) and examined in a transmission electron microscope (JEOL/JEM 2000 EX, Japan). A total of 100 microphotographies were analyzed in this study.
Statistical analysis
Statistical analyses were performed using the Prism 5.0 statistical software package (GraphPad Prism Inc., USA). The data were expressed as mean ± standard error of the mean (SEM), excepting for the variation of body weight, expressed as group means. Statistical comparisons between experimental groups were performed with Mann-Whitney’s non-parametric U-test. All tests used 0.05 as threshold for statistical significance (p < 0.05) and 95% confidence intervals.
RESULTS AND DISCUSSION
The evaluation of clinical signs has been used as main response variable during the development of the present EAE model, which will be used to identify potential therapeutic candidates [21]. It is critical, therefore, to guarantee that the researcher is well trained in the proper procedure for clinical exploration of the animal, according to the selected scale. Weakness in the distal segment of the tail and motor irregularities during walking are the first symptoms of damage to the central nervous system. In order to detect motor irregularities while walking, the mice must be placed onto a flat surface. During normal walking the abdomen of the animals must remain parallel to the surface. Affected animals, however, tend to adopt a crooked stance, and their walking motions are irregular, especially in the hind limbs. Another possibility is to hang the mice from the tail during the clinical evaluation (extension reflex). Retraction of any of the hind limbs to the abdominal region while hanging in this position constitutes one of the initial signs of the disease, and is associated with pathological processes in the spinal motor nerves, including axonal degeneration [22, 23]. Damage to this reflex was used to define the day from which clinical signs of the disease started to appear and to be analyzed using the qualitative scale described in previous sections.
Another useful test is the observation of postural reflexes, performed by turning the animal on its back. Healthy animals turn around onto their feet faster than diseased animals, which may even lose this reflex totally as EAE progresses.
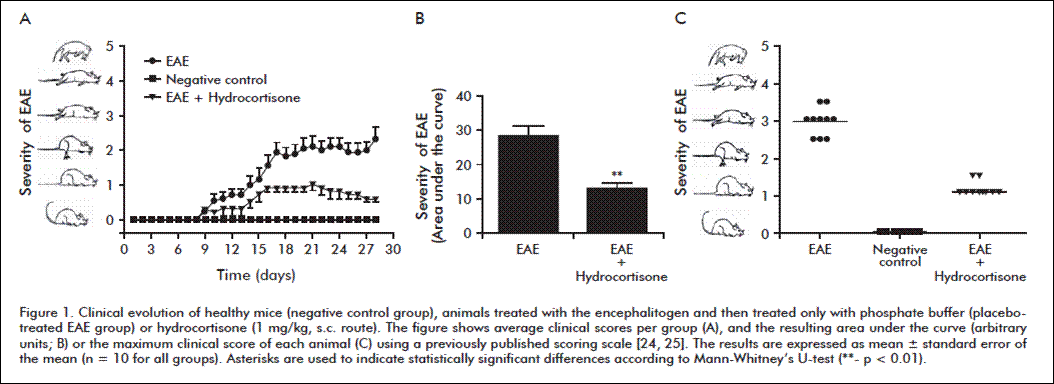
Figure 1A shows average clinical indexes for each experimental group, per day. In diseased animals, clinical signs started to appear by day 8 post-induction. This is a relatively early onset within the range of 7-14 days post-induction described by other authors [26, 27]. The clinical index reached a maximum value of 3.5, evidencing that the experimental procedure successfully induced the disorder. Symptoms of the disorder persisted until the end of the study, coinciding with the chronic progressive form of EAE described by Amor et al. [28].
According to criteria from Vogel et al. [29], a necessary part of the development of an experimental model for a specific disease is the evaluation of the effect exhibited by drugs routinely used in clinical practice against the modeled disease, characterizing the subjacent molecular mechanisms to find out whether they mimic the physiopathological processes taking place in actual patients. Hydrocortisone is one of the steroids that have been used during the last 50 years to treat relapses in MS patients, defined as mild to incapacitating neurological dysfunctions lasting more than 24 hours [30]. The positive effect of hydrocortisone on the clinical symptoms of EAE-affected animals (Figure 1A) was corroborated with statistical comparisons (Figure 1B). Figure 1C shows the maximum indexes reached by each animal on the clinical scale. Animals of the placebo-treated EAE group exhibited high clinical scores evidencing the appearance and progression of the disorder, whereas the animals treated with hydrocortisone improved significantly when compared to the diseased individuals. The results indicate that the model has clinical value, and can be used to establish an adequate correlation for preclinical studies of new drugs against MS.
Body weight is an important variable that provides information related to the degree of neurological deterioration in affected animals. Figure 2 shows how body weight decreases in animals of the placebo-treated EAE group from day 11 of the study. A critical variation takes place between days 12 to 17 post-induction, in close correspondence with the evolution of the symptoms of the disorder. Body weight starts to rebound from day 17 onwards, probably due to compensatory physiopathological mechanisms that exemplify the complexity of this disorder. During some stages, clinical evaluation may not follow or mimic the behavior of molecular parameters, and body weight was therefore explored solely in a descriptive manner. We suggest that body weight constitutes a significant clinical correlate only during the initial period of the disease, when the first clinical signs appear and this variable follows a downward trend. Once the disorder reaches chronicity, the correlation between body weight and clinical signs disappears, and the former follows an unsteady, but decidedly upwards trend. In the negative control group, body weight increased steadily throughout the study, at a rate above that of diseased animals.
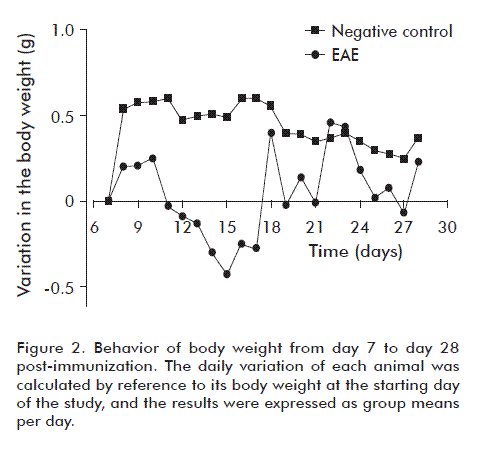
The drop in body weight detected in the placebo-treated EAE group may have a number of causes. One of them is motor deterioration, which may compromise access of the affected individuals to the food placed in their boxes. In this case, this variable would constitute a correlate of disease severity. Another possibility is a decrease in appetite, which has been associated to the effects of leptin in this animal model [31]. Leptin is a hormonal peptide produced by adipocytes that participates in the regulation of energy balance and food intake in the organism [32]. Among its multiple effects, leptin has been shown to inhibit the sensation of hunger when acting upon the hypothalamus, consequently decreasing the ingestion of food, and to stimulate the sympathetic nervous system, thereby increasing metabolic activity and the consumption of energy [32]. It has been shown that blood leptin concentrations increase after the induction of EAE, correlating with the decrease in body weight that takes place during part of the autoimmune process [33].
The evaluation by qPCR of the changes in cytokine profile produced after the induction of EAE, as well as the histological findings of the transmission electron microscopy study, confirmed that the EAE model had indeed been implemented.
According to the results, the diseased animals exhibited increased levels of IL-17 mRNA, in agreement with the data published by other authors working on this model [34]. It was the studies of Langrish comparing IL-12p35-/- and IL-23p19-/- mice that first demonstrated that Th17 cells were responsible for this disorder [35]. Additional evidence concerning the role of Th17 cells on the physiopathology of EAE was obtained in mice lacking the T-bet-/- and STAT6-/- transcriptional factors, which are unable to produce Th1 and Th2 cells, respectively [24]. There is data pointing to Th17 cells as effectors in this disorder [25].
Other effector cytokines (tumoral necrosis factor alfa (TNF-α), IL-1β, IL-6, IFN-γ), as well as markers for natural (Foxp3) and adaptive (transforming growth factor beta, TGF-β) Treg cells showed that EAE-affected animals positively modulated the synthesis of pro-inflammatory effector cytokines, resulting in an increased regulatory response as homeostatic mechanism to restore the effector-regulatory balance of the immune response (Figure 3).
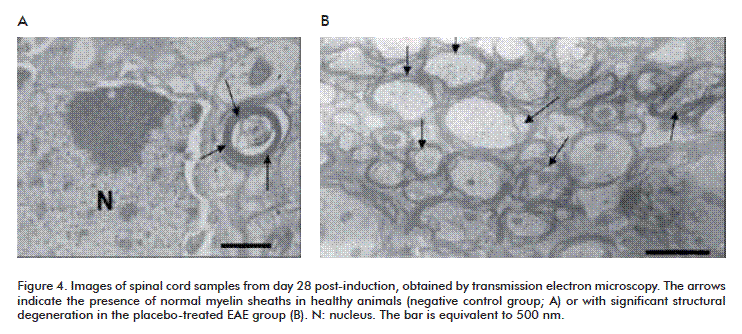
When spinal cord samples were analyzed by transmission electron microscopy, the individuals from the negative control group exhibited compact, dense myelin sheaths and no signs of axonal damage (Figure 4A). In contrast, most axons in animals of the placebo-treated EAE group (Figure 4B) presented interruptions of the continuity of myelin layers. Taking into account that demyelination is one of the physiopathological characteristics of MS [36] and one of its most important pharmacological targets [37], our results confirm that the experimental model we have implemented is a robust tool for mechanistic and preclinical evaluation studies in this field.
CONCLUSIONS
We have implemented a model of chronic, progressive EAE in C57BL6 mice where the first clinical signs appear by day 8 and progressively increase until day 28 post-immunization. The maximum clinical score of the diseased animals reached high values during the development of EAE, and there was correspondence between clinical signs, the results of molecular assays and the observed ultrastructural modifications, which consisted mainly of demyelination and axonal damage at the spinal cord.
REFERENCES
1. Holmoy T, Vartdal F. The immunological basis for treatment of multiple sclerosis. Scand J Immunol. 2007;66(4):374-82.
2. Huynh T. The multiple sclerosis market. Nat Rev Drug Discov. 2010;9(10):759-60.
3. Thomson Healthcare. Red Book: Pharmacy's Fundamental Reference. 112 ed. Montvale, NJ: Thomson Healthcare; 2008.
4. Hohlfeld R, Wekerle H. Immunological update on multiple sclerosis. Curr Opin Neurol. 2001;14(3):299-304.
5. Cassan C, Liblau RS. Immune tolerance and control of CNS autoimmunity: from animal models to MS patients. J Neurochem. 2007;100(4):883-92.
6. Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8(5):337-48.
7. Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med. 2007;13(2):139-45.
8. McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol. 2007;8(9):913-9.
9. Zamvil SS, Steinman L. The T lymphocyte in experimental allergic encephalomyelitis. Annu Rev Immunol. 1990;8:579-621.
10. Zorzella SF, Seger J, Martins DR, Pelizon AC, Sartori A. Resistance to experimental autoimmune encephalomyelitis development in Lewis rats from a conventional animal facility. Mem Inst Oswaldo Cruz. 2007;102(8):931-6.
11. Sewell DL, Reinke EK, Hogan LH, Sandor M, Fabry Z. Immunoregulation of CNS autoimmunity by helminth and mycobacterial infections. Immunol Lett. 2002;82(1-2):101-10.
12. Olfert ED, Cross BM, McWilliam A. Manual sobre el cuidado y uso de los animales de experimentación [Internet]. Estol L, Dugas R, Secretaria Ejecutiva del Consejo Canadiense de Protección de los Animales, editors. Ottawa: Consejo Canadiense de Protección de los Animales [cited 2011 Dec 5]. Available from: http://www.ccac.ca/en_/standards/guidelines/additional/spanish-guide-vol1.
13. Pluchino S, Quattrini A, Brambilla E, Gritti A, Salani G, Dina G, et al. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature. 2003;422(6933):688-94.
14. Esposito M, Ruffini F, Bellone M, Gagliani N, Battaglia M, Martino G, et al. Rapamycin inhibits relapsing experimental autoimmune encephalomyelitis by both effector and regulatory T cells modulation. J Neuroimmunol. 2010;220(1-2):52-63.
15. Peiris M, Monteith GR, Roberts-Thomson SJ, Cabot PJ. A model of experimental autoimmune encephalomyelitis (EAE) in C57BL/6 mice for the characterisation of intervention therapies. J Neurosci Methods. 2007;163(2):245-54.
16. Lech M, Avila-Ferrufino A, Skuginna V, Susanti HE, Anders HJ. Quantitative expression of RIG-like helicase, NOD-like receptor and inflammasome-related mRNAs in humans and mice. Int Immunol. 2010;22(9):717-28.
17. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55(4):611-22.
18. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402-8.
19. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403-10.
20. Spurr AR. A low-viscosity epoxy resin embedding medium for electron microscopy. J Ultrastruct Res. 1969;26(1):31-43.
21. Racke MK. Experimental autoimmune encephalomyelitis (EAE). Curr Protoc Neurosci. 2001;Chapter 9:Unit9 7.
22. Lo AC, Saab CY, Black JA, Waxman SG. Phenytoin protects spinal cord axons and preserves axonal conduction and neurological function in a model of neuroinflammation in vivo. J Neurophysiol. 2003;90(5):3566-71.
23. White SR, Barnes CD. Spinal and spino-bulbo-spinal reflexes in rats with experimental allergic encephalomyelitis. Brain Res. 1975;84(1):123-8.
24. Das J, Ren G, Zhang L, Roberts AI, Zhao X, Bothwell AL, et al. Transforming growth factor beta is dispensable for the molecular orchestration of Th17 cell differentiation. J Exp Med. 2009;206(11):2407-16.
25. Onishi RM, Gaffen SL. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology. 2010;129(3):311-21.
26. Hemmer B, Hartung HP. Toward the development of rational therapies in multiple sclerosis: what is on the horizon? Ann Neurol. 2007;62(4):314-26.
27. Montero E, Nussbaum G, Kaye JF, Perez R, Lage A, Ben-Nun A, et al. Regulation of experimental autoimmune encephalomyelitis by CD4+, CD25+ and CD8+ T cells: analysis using depleting antibodies. J Autoimmun. 2004;23(1):1-7.
28. Amor S, Groome N, Linington C, Morris MM, Dornmair K, Gardinier MV, et al. Identification of epitopes of myelin oligodendrocyte glycoprotein for the induction of experimental allergic encephalomyelitis in SJL and Biozzi AB/H mice. J Immunol. 1994;153(10):4349-56.
29. Vogel HG, Vogel WH, Schölkens BA, Sandow J, Müller G, Vogel WF, editors. Drug discovery and evaluation. Pharmacological assays. 2nd ed. New York: Springer-Verlag; 2002.
30. Burton JM, O'Connor PW, Hohol M, Beyene J. Oral versus intravenous steroids for treatment of relapses in multiple sclerosis. Cochrane Database Syst Rev. 2009(3):CD006921.
31. De Rosa V, Procaccini C, La Cava A, Chieffi P, Nicoletti GF, Fontana S, et al. Leptin neutralization interferes with pathogenic T cell autoreactivity in autoimmune encephalomyelitis. J Clin Invest. 2006;116(2):447-55.
32. Guyton AC y Hall JE. Dietary Balances; Regulation of Feeding; Obesity and Starvation; Vitamins and Minerals. In: Textbook of Medical Physiology. 11th. ed. Pennsylvania: Elsevier-Saunders; 2006. p. 865-80.
33. Sanna V, Di Giacomo A, La Cava A, Lechler RI, Fontana S, Zappacosta S, et al. Leptin surge precedes onset of autoimmune encephalomyelitis and correlates with development of pathogenic T cell responses. J Clin Invest. 2003;111(2):241-50.
34. Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177(1):566-73.
35. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201(2):233-40.
36. Pittock SJ, Lucchinetti CF. The pathology of MS: new insights and potential clinical applications. Neurologist. 2007;13(2):45-56.
37. Mullard A. Success of immunomodulators in MS shifts discovery focus to neuroprotection. Nat Rev Drug Discov. 2011;10(12):885-7.
Received in November, 2011.
Accepted in April, 2012.
Giselle Pentón-Rol. Centro de Ingeniería Genética y Biotecnología (CIGB). Ave. 31 e/ 158 y 190, Cubanacán, Playa, CP 11600, La Habana, Cuba. E-mail: giselle.penton@cigb.edu.cu.

{kind=link}
{kind=link}
{kind=link}
{kind=link}