










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBiotecnología Aplicada
versión On-line ISSN 1027-2852
Biotecnol Apl vol.30 no.4 La Habana oct.-dic. 2013
RESEARCH
Characterization of the IVS17bTA microsatellite marker and six CTFR gene mutations in 21 Cuban families with cystic fibrosis
Caracterización del marcador microsatélite IVS17bTA y seis mutaciones del gen CFTR en 21 familias cubanas con fibrosis quística
Laura González, Teresa Collazo, Yulia Clark, Manuel Gómez, Lídice Reyes
Laboratorio de Biología Molecular, Centro Nacional de Genética Médica, CNGM. Ave. 31 Esq. 146 No. 3102, Reparto Cubanacán, Playa, CP 11400, La Habana, Cuba.
ABSTRACT
Cystic fibrosis is an autosomal recessive disease. Its incidence in Cuba is 1 in 5000 live births. The molecular cause underlying this disease is related to mutations in the regulatory gene encoding the cystic fibrosis transmembrane regulator (CFTR). In this study, the techniques for the study of IVS17bTA microsatellite marker were standardized, and the most frequent mutations in the CFTR gene were also detected for 21 Cuban families. Polymerase chain reaction, agarose and polyacrylamide (plus silver staining) gel electrophoresis techniques were used for both, identification of mutations and microsatellite standardization. Among the 22 Cuban patients, seven were found homozygous, four compound heterozygous and 11 with a single mutation so, 33 chromosomes were molecularly characterized for the 75 %. Twelve allelic variants were found for the IVS17bTA microsatellite; alleles 31 and 7 the most frequent ones. Alleles 31 and 46 were associated to the F508del and R334W mutations, respectively. Among the 21 families, 18 were completely informative for the IVS17bTA microsatellite marker, accounting for 85.7 %. This study helped to complete the diagnosis in those patients in which the responsible mutations in one or both chromosomes had not yet been identified. Besides, we were also able to identify mutations associated with different alleles of the marker without gene sequencing, also helping to decrease the cost of screening.
Keywords: IVS17bTA microsatellite marker, cystic fibrosis, polymerase chain reaction.
RESUMEN
La fibrosis quística es una enfermedad autosómica recesiva. Su incidencia en Cuba es de 1 de cada 5000 recién nacidos vivos. La causa molecular que provoca esta enfermedad son las mutaciones en el gen regulador transmembranal de la fibrosis quística (CFTR). En este estudio se detectaron las mutaciones más frecuentes en 21 familias cubanas y se estandarizaron las técnicas para el estudio del marcador microsatélite IVS17bTA, lo cual constituyó nuestro objetivo principal. Para la identificación de estas mutaciones y la estandarización del microsatélite, se emplearon las técnicas de reacción en cadena de la polimerasa y electroforesis en geles de agarosa y poliacrilamida (tinción con plata). De los 22 pacientes, se encontraron 7 homocigóticos, 4 heterocigóticos compuestos y 11 con una sola mutación, por lo que se caracterizaron molecularmente 33 cromosomas, lo que representa el 75 %. En el estudio del microsatélite IVS17bTA se encontraron 12 variantes alélicas, los alelos más frecuentes fueron el 31 y el 7. El alelo asociado a la mutación F508del fue el 31 y a la mutación R334W fue el 46. De las 21 familias analizadas, 18 resultaron completamente informativas para el marcador, lo que representa el 85.7 % Este estudio ha ayudado a completar aún más el diagnóstico en los pacientes en los que aún no se han identificado las mutaciones responsables en ambos cromosomas o en uno de ellos. Además se han podido identificar mutaciones asociadas a diferentes alelos del marcador, sin necesidad de secuenciar el gen, lo que aumentaría el costo.
Palabras clave: marcador microsatélite IVS17bTA, fibrosis quística, reacción en cadena de la polimerasa.
INTRODUCTION
Cystic fibrosis (CF) is one of the most common genetic diseases in Caucasian populations, becoming a worldwide health problem. Cuba has reported an incidence of approximately one per 5000 live births [1].This multisystemic disease is clinically characterized by pancreatic insufficiency, progressive lung disease and high concentration of electrolytes in sweat. CF patients suffer from physical and psychological limitations. The clinical treatment to overcome this disease is often lengthy and costly, because of the required antibiotics and pancreatic enzyme replacement, proper nutrition and long-term hospitalization.
The inheritance pattern of CF is autosomal recessive, caused by mutations in the Cystic Fibrosis Transmembrane Regulator (CFTR) gene, which is located on chromosome 7 region 7q31. This gene of approximately 250 kDa contains 27 exons encoding for a 1480 amino acids protein with a molecular weight of approximately 170 kDa. [2]. More than 1800 mutations in the CFTR gene have been detected [3].
In Cuba, CF diagnosis is done based on the patient’s clinical picture, joined to the positive result of electrolytes test in sweat. Besides, six mutations are detected frequently in our country [4, 5]: F508del (37.9 %), G542X (6.9 %), R334W (5.2 %), R553X (2.2 %), R1162X (2.0 %) and 3120+1G→A (1.3 %). However, the coverage of the affected chromosome reaches only 55 % with the detection of these six mutations. Therefore, a broader range of diagnostic, molecular markers like IVS17bTA, IVS8CA and IVS17bCA microsatellites are required.
Microsatellites are DNA sequences containing variable mononucleotide, dinucleotide (TG) or trinucleotide (CAA)n repeats, with alleles constituted by different numbers of units [6]. They are used to screen and characterize mutations, in gene evolutionary studies and indirect disease diagnosis [7].
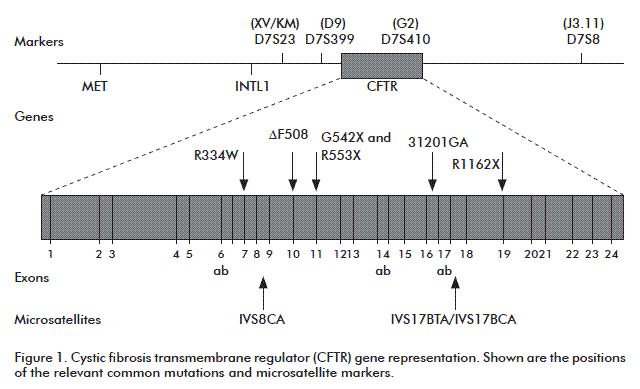
CFTR microsatellites are located in intronic regions (Figure 1). Particularly, the IVS17bTA microsatellite is located in intron CFTR 17b, of which 30 alleles have been identified bearing 7 to 56 TA repeat units [8]. The most common alleles bear 7, 30 and 31 dinucleotide units, for 0.22, 0.19 and 0.12 frequencies, respectively, among non-fibrocystic chromosomes [9]. This microsatellite is highly heterozygous and allows reaching information close to 99 % when combined with the most common mutations, according to studies in Caucasian populations [8].
VS17bTA is globally reported as the most informative among the three microsatellite markers mentioned, due to a high number of alleles. For this reason, our work was based on standardization and the analysis of this particular microsatellite in Cuban fibrocystic patients.
MATERIALS AND METHODS
Study subjects
Sixty-four individuals from 21 families were studied, 22 suffering from CF. Diagnoses were made according to patients’ clinical features and the sweat electrolyte test results. The patients were diagnosed by the National Cystic Fibrosis Cuban Commission (Cuban Ministry of Health, MINSAP).
Mutations F508del, G542X, R1162X, R553X, 3120+1G→A and R334W were evaluated in all the individuals.
DNA extraction
DNA was obtained from a 10-mL peripheral blood sample using 56 mg/mL ethylenediaminetetraacetic acid (EDTA) as anticoagulant. DNA extraction was carried out by the salting-out method described by Miller et al. in 1988 [10]. Once the DNA was isolated, samples were coded and stored until further use.
Detection of mutations
F508del, G542X and R1162X mutations were detected by the amplification refractory mutation specific (ARMS) method described by Newton et al.[11]. Each reaction contained: 100 ng of DNA, 1 U of Taq DNA polymerase (Invitrogen, USA), 0.85 pmol/L of each primer (Biosource, USA), 0.1 mM dNTP, 1.0 mM MgCl2 in a final volume of 25 µL. In all cases, internal amplification controls were also used. The amplification reaction was carried out according to the following steps: denaturing at 94 °C for 5 min and thereafter Taq DNA polymerase (Promega, USA) was added, followed by 29 cycles of denaturating at 94 °C for 1 min, annealing at 60 °C for 1 min, and extension at 72 °C for 90 s, followed by a final extension cycle at 72 °C for 5 min. The DNA fragments obtained were run at 250 V for 25 min in a 2 % agarose gel containing ethidium bromide and visualized on an UV light transilluminator.
Mutations 3120+1G→A, R553X and R334W, were detected by polymerase chain reaction (PCR), followed by enzyme restriction analysis specific for each mutation and agarose gel electrophoresis.
To detect 3120+1G→A, the following conditions were established per reaction: 100 ng DNA, 1 U of Taq DNA polymerase (Invitrogen, USA), each primer at 6.5 pmol/µL (Biosource, USA), 0.1 mM dNTP, 2.5 mM MgCl2 in a final reaction volume of 25 µL. The amplification reaction consisted of denaturing at 94 ºC for 5 min, followed by 35 cycles of denaturing at 94 °C for 1 min, annealing at 57 °C for 1 min, extension at 67 °C for 1 min; and a final extension cycle at 67 °C for 5 min. The amplified product was further digested with 20 U of BstN I (Bio Labs, USA) per sample in a final volume of 35 µL, at 60 °C for 4 h.
The R334W mutation was analyzed under the following conditions per reaction: 100 ng DNA, 1 U Taq DNA polymerase (Invitrogen, USA), 6.5 pmol/µL (Biosource, USA) for each primer, 0.1 mM dNTP, 1.5 mM MgCl2, in a reaction volume of 25 µL. The amplification reaction consisted of denaturing for 3 min, followed by 35 cycles of repetitions consisting of 20 s of denaturing at 94 °C, annealing at 55 °C for 30 s, extension at 74 °C for 30 s and a final extension cycle at 74 °C for 5 min. The amplified product was digested with 25 U of the restriction enzyme Hpa II (Promega, USA) in a final volume of 35 µL, at 37 °C for 3 h.
The digested amplification products for the 3120+1G→A and R334W mutations were electrophoresed in 2 % agarose gels containing ethidium bromide and bands were visualized under UV light.
In the case of the R553X mutation, amplification reaction conditions were: 100 ng DNA, 1 U Taq DNA polymerase (Invitrogen, USA), each primer at 6.5 pmol/µL (Biosource, USA), 0.1 mM dNTP, 1.5 mM MgCl2 in a final volume of 25 µL. The reaction mixtures were denatured at 94 ºC for 5 min, followed by 30 cycles of denaturing at 94 °C for 30 s, annealing at 55 °C for 30 s, extension at 72 °C for 1 min, and a final extension cycle at 72 °C for 5 min. The amplified product was digested with 25 U of Hinc II (Bio Labs, USA) to a final volume of 35 µL, at 37 °C for 3 h. The digested product was subsequently run on a 3 % agarose gel and visualized on a UV light transilluminator.
Microsatellite marker analysis
The microsatellite IVS17bTA was amplified by PCR under the following conditions per reaction: 100 ng DNA, 1 U of Taq DNA polymerase (Invitrogen, USA), 5 pmol/L of each primer (Biosource, USA), 0.1 mM dNTP, 1.5 mM MgCl2 in a final volume of 25 µL. The amplification reaction was carried out by an initial denaturing step at 94 ºC for 4 min, followed by 30 cycles with the following steps: denaturing at 94 °C for 30 s, annealing at 50 °C for 30 s, extension at 72 °C for 30 s; and a final cycle of extension at 72 °C for 7 min. PCR primer sequences are shown in table 1. Amplification products were verified by electrophoresis in a 2 % agarose gel, with bands ranging 202-294 bp, according to the obtained allelic variants.
The products were further subjected to 12.5 % polyacrylamide gel electrophoresis, with a migration time from 4 to 5 h at 150 V and a temperature of 15 °C. Subsequently, DNA was visualized through gel silver staining with the PlusOne DNA Silver Staining Kit (Amersham Biosciences, USA, 2002) according to manufacturers’ instructions.
RESULTS
Among the 64 tested people, 22 were suffering from CF and 42 were carriers. From these last, only 33 carried the identified mutations.
In 11 patients two mutated alleles were present, with the other 11 carrying only one mutated allele. In total, heterozygous, homozygous and heterozygous compound alleles were carried by 55 people.
Table 2 shows the results of detected mutations in the 21 families under study. Mutations R1162X, R553X and 3120+1G→A were not present in the analyzed families.

Among the 22 studied patients, seven were homozygous (six with F508del and one with G542X), four were compound heterozygous (two with F508del/R334W, one with F508del/G542X and one with G542X/R334W) and 11 carried a single mutation (F508del or R334W), accounting for 33 molecularly characterized chromosomes (75 %). Figure 2 shows the frequencies of the IVS17bTA marker alleles identified in the 64 individuals; 12 out of the 30 allelic variants described in other Caucasian populations were found [8].
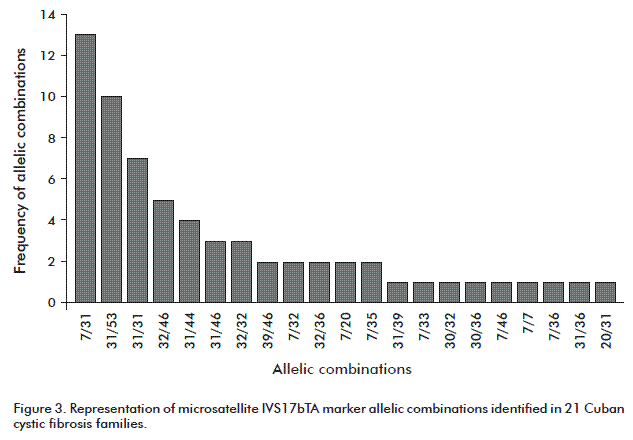
The most frequent alleles in descending order in the 64 studied individuals were 31, 7, 46, 32 and 53. The less frequent ones were 33, 35, 20 and 30.
The IVS17bTA microsatellite marker frequencies for the allelic combinations identified in the study are shown in figure 3. The most common allelic combinations in descending order were: 7/31, 31/53, 31/31 and 32/46. It was found that the IVS17bTA microsatellite marker allele 31 is the most common in the Cuban population, as in Spain and other Caucasian populations [12]. These data are taken as reference because of the genetic background contribution of the Spanish population to the Cuban genome.
Figure 4 shows the frequency of the IVS17bTA microsatellite marker alleles found in the 22 CF patients; the most frequent in descending order were 31, 7, 46, 32 and 53. The less frequent ones were 35, 39 and 44.
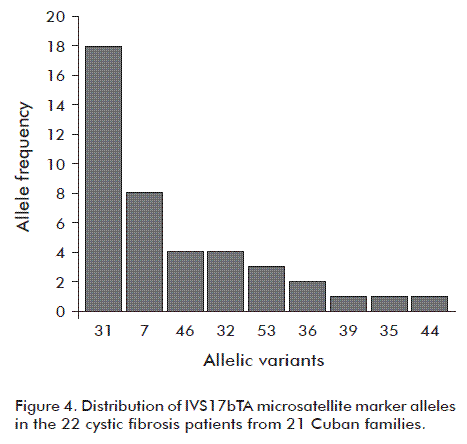
Similar to European populations [12], it was observed that the IVS17bTA microsatellite marker alleles 31 and 32 were the most common in the Cuban CF patients.
The allelic combinations of the IVS17bTA microsatellite marker identified in the 22 CF patients are shown in figure 5, the most common: 31/31, 7/31, 31/53 and 32/46. Conversely, the less frequent combinations were 31/46, 32/36, 7/32, 39/46, 7/7, 7/35 and 31/44.
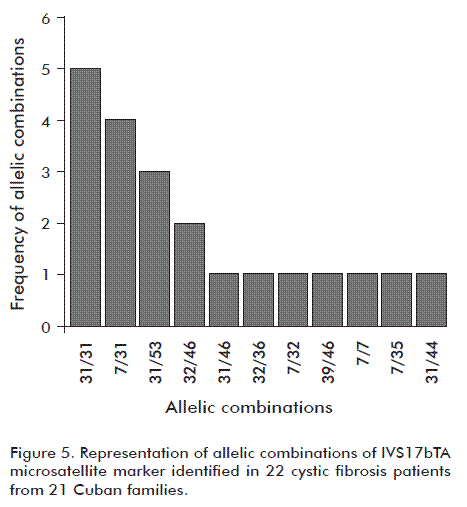
An association analysis of identified CTFR mutations and the IVS17bTA microsatellite allele marker showed that allele 31 was associated to mutation F508del, as found in 34 individuals. For the G542X mutation, there were six individuals carrying it together with IVS17bTA allele 32. These results are in agreement with those reported worldwide and open the search for these mutations in the Cuban population.
In addition, we studied the heterozygosity of the IVS17bTA microsatellite marker in the 64 individuals. There were 55 heterozygous and 9 homozygous individuals for the marker alleles, for 85.93 % heterozygosity. This pointed out this microsatellite as highly informative. The informativeness analysis performed in the 21 families found that 18 (85.7 %) were completely informative and only three semi-informative.
DISCUSSION
The use of genetic markers is an alternative to detect the transmission of mutated alleles in population groups, despite the knowledge on point mutations responsible for the disease. The discovery of the polymorphic markers aim of the present study, closely linked to the CFTR gene within the affected gene, is a major breakthrough to provide indirectly prenatal and carriers CF diagnosis. Additionally, presymptomatic screenings can be run in newborns, providing the opportunity to implement prevention, to apply a more specific treatment and to improve the quality of life of these patients.
Considering the origin of the Cuban population, the most common mutations reported for the CFTR gene in Spain (F508del, G542X, R1162X, R334W and R553X) and Africa (3120+1G→A) were searched in Cuban patients. As in most populations around the world, the F508del mutation was found as the predominant cause for CF in patients showing severe clinical symptoms.
Sixty-nine percentage of the subjects and seventy-five percentage of CF chromosomes were genotyped, demonstrating a high molecular heterogeneity of the disease in the analyzed sample.
The knowledge on the association of the IVS17bTA microsatellite marker alleles to the most frequent CFTR mutations can extend the diagnostic possibilities. Studies carried out in the Spanish population showed that allele 31 of this microsatellite marker is associated with F508del mutation, and alleles 46 and 47 with R334W [12]. Also in other countries near the Mediterranean and the British Isles, the F508del mutation appears associated with allele 31 of the same microsatellite marker [10].
Noteworthy, this is the first study in Cuba standardizing PCR, and agarose or polyacrylamide gels electrophoresis conditions for the VS17bTA microsatellite marker. With the identification of VS17bTA alleles and its high heterozygosity in the 64 individuals tested, 85.7 % families were informative. This result highlights the informativeness of IVS17bTA, described as the most informative of the three micro satellite markers located in the CFTR gene. Figure 6 shows a family regarded as informative for the marker and an example of prenatal diagnosis.

A family is fully informative when parents are heterozygous for those marker alleles, since in each parent it is possible to determine which allele segregates with the disease and so, this allows further study of the linkage. This analysis is only relevant if the family has at least one affected member. In the example, the marker alleles that segregate with the disease are 31 in the mother and 53 in the father. It was previously known from the family history that alleles 31/53 were associated with the disease. In this way, the prenatal diagnosis can be established to identify the fixed possible outcomes (Figure 6).
In summary, the IVS17bTA microsatellite marker is an excellent indicator for genetic linkage studies in families with CF, both for carrier detection and prenatal diagnosis. The implementation of IVS17bTA studies widens the molecular diagnostic possibilities in fibrocystic Cuban families.
REFERENCES
1. Collazo T, Piloto Y, Clark Y, Boffil AM, Gómez M, Hernández Y. Detección de mutaciones en pacientes cubanos con fibrosis quística. Biotecnol Apl. 2008;25(4):345-9.
2. Scriver CR, Beaudet AL, Sly GW, Valle D, editors. The metabolic and molecular bases of inherited diseases. 7th ed. Vol. 3. New York; Mc Graw-Hill; 1995.
3. Cystic Fibrosis Mutation Database (CFMDB) [Internet]. Ontario: Cystic Fibrosis Consortium. c2011 [cited 2013 May 9]; Available from: http://www.genet.sickkids.on.ca/cftr/StatisticsPage.html
4. Collazo T, Bofill AM, Clark Y, Hernandez Y, Gomez M, Rodriguez F, et al. Common mutations in Cuban cystic fibrosis patients. J Cyst Fibros. 2009;8(1):47-9.
5. Collazo T, López I, González L, Clark Y, Piloto Y, Gómez M, et al. Estudio molecular de la fibrosis quística en Cuba. In: Memorias Convención Internacional de Salud Pública. Cuba Salud 2012. La Habana 3-7 de diciembre de 2012, Cuba. La Habana: Ministerio de Salud Pública; 2002.
6. Weber JL, May PE. Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am J Hum Genet. 1989;44(3):388-96.
7. Estivill X, Morral N, Casals T, Nunes V. Prenatal diagnosis of cystic fibrosis by multiplex PCR of mutation and microsatellite alleles. Lancet. 1991;338(8764):458.
8. Visich AA, Barreiro CZ, Chertkoff LP. Caracterización de tres microsatélites del gen de fibrosis quística en familias argentinas. Medicina (Buenos Aires). 2001;61(1):23-7.
9. Zielenski J, Rozmahel R, Bozon D, Kerem B, Grzelczak Z, Riordan JR, et al. Genomic DNA sequence of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics. 1991;10(1):214-28.
10. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215-8.
11. Newton CR, Graham A, Heptinstall LE, Powell SJ, Summers C, Kalsheker N, et al. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 1989;17(7):2503-16.
12. Morral N, Bertranpetit J, Estivill X, Nunes V, Casals T, Gimenez J, et al. The origin of the major cystic fibrosis mutation (delta F508) in European populations. Nat Genet. 1994;7(2):169-75.
Received in October, 2012.
Accepted in May, 2013.
Teresa Collazo. Laboratorio de Biología Molecular, Centro Nacional de Genética Médica, CNGM. Ave. 31 Esq. 146 No. 3102, Reparto Cubanacán, Playa, CP 11400, La Habana, Cuba. E-mail: tcollazo@infomed.sld.cu.


{kind=link}
{kind=link}
{kind=link}
{kind=link}