










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBiotecnología Aplicada
versión On-line ISSN 1027-2852
Biotecnol Apl vol.31 no.2 La Habana abr.-jun. 2014
RESEARCH
DNA damage and repair capacity in patients with neurofibromatosis type 1
ADN dañado y capacidad de reparación en pacientes con neurofibromatosis tipo 1
Reinaldo Gutierrez1, Judith Pupo1, Gretel Riverón1, Ana M González2, Anamarys Pandolfi1, Aimara de Armas1, Mildrey Cásido1, Iris Rojas1
1 Centro Nacional de Genética Médica, CNGM. Calle 146 No. 3102. Playa, CP 10600, La Habana, Cuba.
2 Centro Provincial de Genética, Holguín, Cuba.
ABSTRACT
Neurofibromatosis type 1 (NF1) is an autosomal dominant disorder which displays considerable inter- and intra-familial variability in phenotypic expression. NF1 is characterized particularly by café-au-lait spots and fibromatous tumors of the skin. In this study, the comet assay was used to evaluate levels of basal single strand breaks, H2O2 oxidation-induced DNA damage, and repair capacity in lymphocytes of NF1 patients compared to healthy control subjects. No significant differences in DNA damage were observed between controls and patients (p > 0.05), but DNA repair capacity was significantly slower in NF1 patients (p < 0.05). It suggests less efficient DNA repair capacity may be associated with NF1 disease. Using this assay we could identify individuals with poor repair capacity who would be good candidates for intensive follow-up and screening.
Keywords: DNA repair, neurofibromatosis, peripheral blood lymphocytes, comet assay, oxidative damage.
RESUMEN
La neurofibromatosis tipo 1 (NF1) es una enfermedad autosómica dominante que muestra una gran variabilidad inter e intrafamiliar en la expresión fenotípica. Se caracteriza sobre todo por manchas de color café con leche y tumores fibromatosos de la piel. En este estudio, se utilizó el ensayo de cometa para evaluar los niveles basales de roturas de simple cadena, la oxidación inducida por H2O2 en el ADN, y la capacidad de reparación en los linfocitos de los pacientes con NF1 en comparación con sujetos sanos. No se observaron diferencias significativas en el daño de ADN entre los controles y los pacientes (p > 0.05), pero la capacidad de reparación del ADN fue significativamente más lenta en los pacientes con NF1 (p < 0.05). Esto sugiere que la capacidad de reparación del ADN menos eficiente puede estar asociada con el desarrollo y evolución de la enfermedad. Con la utilización de este ensayo podríamos identificar a los individuos con la capacidad de reparación disminuida, los cuales serían buenos candidatos para un seguimiento intensivo.
Palabras clave: reparación del ADN, neurofibromatosis, linfocitos de sangre periférica, ensayo cometa, daño oxidativo.
INTRODUCTION
Neurofibromatosis type 1 (NF1, OMIM# 162200) is a common autosomal dominant disorder affecting one in 3500 individuals. It is caused by deletion or point mutation of NF1, a tumor suppressor gene mapping to the chromosomal region 17q11.2. About half of the NF1 cases are caused by de novo mutations [1-3].
The main clinical features of the disease are café-au-lait spots, freckling of the axillary or inguinal region, iris Lisch nodules, and cutaneous neurofibromas, the incidence and the number of which differ appreciably from one patient to another [4]. Approximately one third of NF1 patients have plexiform neurofibromas which between 10-15 % of them transform into Malignant Peripheral Nerve Sheath Tumours (MPNSTs) [5]. However, NF1 is notable for its extreme phenotypic variability both inter- and intra- familial. Allelic heterogeneity of the NF1 gene may be one of the factors explaining the great clinical variability of the disease. Diverse mutations have been reported in NF1, but no genotype-phenotype relationships have been established except that large deletions encompassing the whole NF1 gene and its flanking chromosomal regions tend to be found in those with a more severe expression The main clinical features of the disease are café-au-lait spots, freckling of the axillary or inguinal region, iris Lisch nodules, and cutaneous neurofibromas, the incidence and the number of which differ appreciably from one patient to another [4]. Approximately one third of NF1 patients have plexiform neurofibromas which between 10-15 % of them transform into Malignant Peripheral Nerve Sheath Tumours (MPNSTs) [5]. However, NF1 is notable for its extreme phenotypic variability both inter- and intra- familial. Allelic heterogeneity of the NF1 gene may be one of the factors explaining the great clinical variability of the disease. Diverse mutations have been reported in NF1, but no genotype-phenotype relationships have been established except that large deletions encompassing the whole NF1 gene and its flanking chromosomal regions tend to be found in those with a more severe expression.
The ability to repair DNA damage is strongly associated with risk of cancer and other diseases such as neurodegenerative inflammatory disorders. Repair of DNA damage plays an essential role in cell survival and the maintenance of genomic stability [9]. Allelic variations in genes involved in DNA repair pathways can alter an individual’s ability to repair DNA damage, resulting in increased sensitivity to exogenous and endogenous agents and greater susceptibility to mutations and genetic instability [10]. In consequence, this would center attention on factors involved in DNA repair as possible modifiers of the NF1 phenotype, with detection of such phenotypic modifiers having potential prognostic value.
Particularly, DNA damage induced by reactive oxygen species (ROS) may lead to single- or double-strand breaks, point and frame-shift mutations and larger-scale chromosomal abnormalities [11]. Molecular oxygen reaction products induce point mutations, deletions and gene amplification and rearrangement in mammalian cells, which may result in proto-oncogene activation and/or tumor suppressor gene inactivation [12]. Among more than 30 different products of modified DNA by oxidative stress, 8-oxo-7,8-dihydroguanine ( 8-oxoGua) is the most studied mutagenic lesion. This lesion induces an increased frequency of spontaneous G:C or T:A transversion mutations. The oxidative DNA lesion 8-oxoguanine is recognized by the specialized repair enzyme 8-oxoguanine DNA glycosylase (hOGG1). This enzyme can be used as specific tool for identification of oxidized guanine bases, as it reveals these lesions as single strand breaks that can be detected using the single cell gel electrophoresis or comet assay [13-15].
Various biomarkers have been used to determine cellular DNA damage in NF1; cytogenetic measurements include chromosomal aberrations, micronuclei and sister chromatid exchanges [16, 17]. Additionally, the comet assay technique is recognized among the most rapid, simple and sensitive methods available for measuring DNA strand breaks with a small number of cells [18, 19]. The alkaline comet assay resolves break frequencies up to a few thousand per cell, so the distances between breaks are in the order of 109 Da. To examine 8-OhdG levels by this technique, DNAs can be incubated with hOGG1, a commercial endonuclease that generates additional breaks at sites containing 8-oxo-dGua, and by comparing the DNA migration in enzyme-treated and -untreated slides, quantitation can easily be made [20, 21].
MATERIALS AND METHODS
Study subjects
Thirty NF1 patients were enrolled (15 men and 15 women; age as mean ± SD: 24.9 ± 8.2 years). All the subjects were diagnosed to have NF1 based on standard diagnostic criteria at the Juan Manuel Márquez Pediatric Hospital and Hermanos Ameijeiras Clinical Hospital, both in Havana, Cuba. Medical histories were obtained and physical examinations were performed to all the NF1 individuals enrolled. The control group comprised 30 healthy subjects (10 men and 20 women; age: 35.2 ± 8.8 years) from Havana. Exclusion criteria for all subjects were chemotherapy or radiotherapy, infections, and blood transfusion in the previous month. After agreement and signing the informed consent, all participants donated 5 mL of venous blood and completed a questionnaire that provided detailed information on occupational exposures, family history of cancer, medications, reproductive history, and past treatments for noncancer conditions. There were no age and gender restrictions for study eligibility. All the controls and patients were non-smokers.
The laboratory and questionnaire data were coded, entered and verified; neither the laboratory nor the data entry personnel had knowledge of the subjects’ case-control status. Written informed and educated consent was obtained from each patient or healthy volunteers and from parents of all children before entering into this patient-control study. This study was conducted according to the guidelines laid down in the Declaration of Helsinki [22] and approved by the ethics committee of the National Centre of Medical Genetics, Havana, Cuba.
Peripheral blood lymphocyte isolation
Heparinized blood samples from the NF1 patients and control subjects were protected from light, put on ice, and processed within 4 h of collection in the Oxidative Stress Laboratory at the National Centre of Medical Genetics, Havana, Cuba. Lymphocytes were isolated using standard Ficoll-Histopaque method. Briefly, 5 mL of whole blood from each subject was layered over 5 mL of Histopaque-1077 (Sigma Aldrich Co., St. Louis, MO) at 4 ºC and centrifuged at 1500 rpm for 30 min. The mononuclear cells were removed from the interphase, washed twice with cold (4 ºC) phosphate buffered saline (PBS), pH 7.2, and centrifuged at 1500 rpm for 10-15 min. Cells were re-suspended in 1 mL of cold PBS. Manual cell counts and the cell membrane integrity were determined by Trypan Blue solution 0.4 % and the cell suspension was adjusted with PBS to 1 × 106 cells/mL.
DNA damage assessment
Constitutive or endogenous DNA damage as pre-existing single strand breaks was assessed by the comet assay [20] with some modifications. Two slides per each patient and control and two gels per slide (i.e., four gels per patient and control) were
prepared. Briefly, 50 µL of each cell suspension (estimated to contain approximately 1000 cells) were added to 75 µL of 1 % low melting point agarose solution made in PBS buffer at 37 ºC), gently mixed, and the mixture was immobilized on a microscope slides which had previously received a layer of 0.5 % low melting point agarose. When the gel had set, the slides were placed in freshly prepared ice-cold lysis solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris-HCl, and 1 % Triton X-100 with 10 % DMSO, pH 10) to remove cell proteins, leaving DNA as ‘nucleoids’. To allow for DNA denaturation and unwinding and the exposure of the alkali-labile sites, slides were kept for 25 min in a horizontal electrophoresis chamber without power that was filled with freshly prepared alkali buffer (0.3 M NaOH and 10 mM EDTA at pH > 13.0) at 4 °C. After the unwinding, DNA was electrophoresed at 0.8 V/cm and 300 mA for 25 min; all these steps were carried out in subdued light. Finally, the slides were washed three times in neutralizing buffer (0.4 M Tris, pH 7.5) to remove alkali and detergents, and were stained using a silver staining protocol [19]. Slides were: a) fixed for 10 min in a solution containing 15 % trichloroacetic acid, 5 % zinc sulphate heptahydrate, and 5 % glycerol; b) washed three times with deionized water; c) placed back-to-back in a horizontal staining jar; d) stained for 35 min in dark conditions with shaker using 75 mL of freshly prepared stain solution composed by 34 mL of vigorously mixed stock solution B (0.1 % ammonium nitrate, 0.1 % silver nitrate, 2.5 % tungstosilicic acid, 0.15 % formaldehyde, v/v) and 66 mL of stock solution A (5 % sodium carbonate); e) washed three times with deionized water; f) immersed 5 min in a stop solution (acetic acid 1 %); and g) slides were air-dried.
Induced DNA damage and repair
A modification of the basic alkaline comet assay was introduced to test the cells’ response and their capacity to repair after a controlled in vitro oxidative challenge. This was induced by exposure to 200 µmol/L hydrogen peroxide (H2O2, made up in PBS), for 5 min at 4 ºC. Some of the challenged cells were washed and then embedded in agarose and run through the comet assay as described above, to measure its resistance to challenge, while some of the challenged cells were used to assess DNA repair. This last was done by resuspending the washed cells in RPMI 1640 medium containing 20 % fetal calf serum, and incubating the cells at 37 ºC for 90 min, which were further placed on ice to stop DNA repair, and embedded in agarose and the comet assay run. The efficacy of DNA repair was taken as the relative difference between DNA damage immediately after challenge and after 90 min of repair.
DNA repair enzyme treatment
In order to examine basal levels of oxidation-induced damage in DNA, nucleoids embedded in agarose were incubated with 50 µL hOGG1 in buffer (20 mM Tris-HCl, 1 mM EDTA, 1 mM dithiothreitol, 100 µg/mL bovine serum albumin) at 0.08 U per gel, for 45 min at 37 ºC. hOGG1 is an endonuclease that recognizes lesions 8-oxo-dGuo and creates single strand breaks at those sites [23]. For each gel treated with hOGG1 there was a gel incubated in parallel, but with buffer alone. The number of DNA oxidation-induced lesions was expressed as the difference between the enzyme- and the buffer- treated gels.
Scoring of DNA damage
For the aim of evaluating the degree of damage, comet images were scored visually using an optical microscope. Slides were in duplicates for each subject’s sample in all versions of the comet assay used. Two hundred cells (100 cells from each slide replicate) were scored using arbitrary units of damage according to the intensity and length of the comet tail. A 0-4 scale was used, with 0 indicating no damage (all DNA within the nucleoid head and no migration of DNA into the gel) through 4 representing maximal damage. The scores for the 50 nucleoids scored on each gel were summed into a damage index, ranging from 0 (all cells with no damage) to 400 (all cells with maximal damage). The number of comets in each category was counted and average DNA damage in the case of strand breaks was expressed as arbitrary units, which is related to the percentage of DNA in the tail. Slides were analyzed under blind conditions.
To assess sensitivity to H2O2, the induced damage (IND) was calculated as the damage score in the H2O2-stressed cells and the damage score of cells from the same subject but without H2O2 exposure. The repair capacity was calculated as the percentage of H2O2-induced DNA damage remaining after 90 min repair time of stressed cells % RD in relation to the induced damage in cells immediately after exposure to H2O2 [24, 25].
Statistical analysis
The values of the comet assay were expressed as mean ± standard error of the mean. The statistical analyses were performed by the nonparametric Mann-Whitney U-test, since data showed no normal distribution. A p value lower than 0.05 was considered as significant. All the analyses were performed using the Statistica® software version 8 (StatSoft Inc.).
RESULTS AND DISCUSSION
Our study was performed in the course of a multicenter study in Havana about DNA damage in patients with genetic deficiency in tumors suppressor genes. In this study, DNA damage and repair testing was offered to children showing no severe clinical features characteristic of the disease who fulfilled the internationally established minimum clinical criteria in NF1 disease. NF1 group had approximately 25 % higher baseline DNA damage than the control subjects, though this fell just short of statistical significance (p = 0.053), while the hOGG1-sensitive sites were very similar in the two groups. The controls had similar mean H2O2-induced DNA damage compared with cases (198.21 ± 9.94 versus 203.18 ± 10.90; p > 0.05) (Table). The DNA repair percentage of residual DNA damage among cases and controls are summarized in the table. Comparing cases and controls revealed a high significant percentage of residual DNA damage in the controls (74 ± 0.27 versus 45.21 ± 6.13; p = 0.00041), thus reflecting efficient repair capacity compared to the cases.
We dichotomized the repair DNA by first percentile and third percentile of controls using the 25th percentile of the controls as the cutoff point. Patients with results within the 25 to 75 % range of controls were considered to show a ‘normal’ cellular repair to H2O2 exposition. Among the NF1 cases, approximately 71 % fell into the poor repair category (less than 25 % repair of induced damage after incubation), and none of the NF1 showed repair higher than 75 % of the induced damage. However, healthy subjects had a more effective repair capacity, with 76 % showing repair above 75 % of the induced damage at the 90 min incubation (Figure).
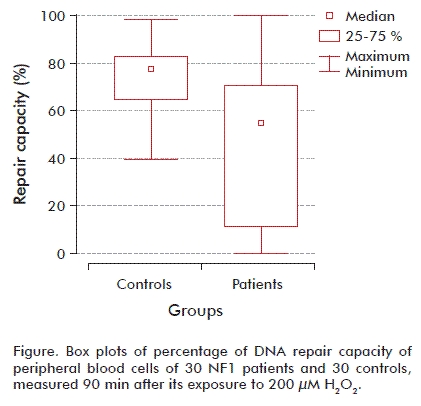
A large number of Mendelian inheritance genetic disorders display considerable inter- and intra-familial variability in phenotypic expression. It is now increasingly apparent that genetic modifiers, distinct from the disease locus itself, have a considerable role to play in phenotypic variance.
In that sense, DNA repair, a major mechanism for maintaining genome integrity and preventing mutations, has evolved into specific DNA repair pathways to repair different types of DNA damage, and to maintain genomic integrity. In the human genome more than 130 genes have been found to be involved in these DNA repair systems [9, 10]. Thus, before launching expensive and time-consuming genetics studies to identify these genetic modifiers, it is important to make sure that they really exist and that environmental factors or other do not suffice to explain this phenotypic variability. The idea that each neurofibroma would result from an independent somatic mutation event, regardless its origin from the same or different patient, was examined by Wiest et al. [26]. They performed a mutational screen of 33 neurofibromas from a mother and her daughter, both with NF1. Tumors from those two patients exhibited a high percentage of small mutations, and the authors proposed that functional variants of DNA repair genes modulate the frequency of second hits of the NF1 gene. Additional support for a role of DNA repair genes as modifiers in NF1 comes from the observation that mutations in DNA mismatch repair (MMR) genes, in the homozygous state, predispose individuals to NF1 [27, 28].
Several children identified as harboring homozygous germline MLH1 or MSH2 mutations, with consequent deficiencies in DNA mismatch repair, developed hematological malignancies at an early age, and exhibited clinical features of de novo NF1 [30-32]. Inactivation of the NF1 gene in MMR-deficient cells may be an early critical step in malignant progression [31]. In Nf1+/− mice, Gutmann and colleagues [32] found that a deficiency in MMR (Mlh1−/−) significantly accelerated myeloid leukemogenesis, with concomitant microsatellite instability and loss of neurofibromin expression in the tumors analyzed.
On the other hand, germline gene alterations play a significant role during malignant transformation of progenitor glial cells. In glioma patients have been demonstrated that germline p53 mutations are frequent in patients with multifocal glioma, gliomas and another primary malignancy [33]. Similarly, germline mutations in DNA repair genes BRCA-1 and BRCA-2 significantly increase the risk of developing multifocal glioblastoma [34]. Some studies suggest that reduced expression of MMR genes is frequent in human gliomas, and aberrant expression of more than one MMR gene may be associated with an increased risk of second primary malignancies in glioma patients [35]. Several studies that examined both spontaneous and induced chromosome instability in lymphocyte cultures suggested that chromosome instability can be detected in the peripheral blood lymphocytes of glioma patients and it may be a marker for identifying individuals at risk [36].
Another factor identified as involved is poly (ADP-ribose) polymerase-1 (PARP-1), an enzyme involved in DNA repair regulation. PARP-1 interaction with NF-κB has been identified as a major factor regulating macrophage and microglial activation. PARP-1 gene deficiency prevents the morphological changes associated with microglial activation, and suppresses microglia release of proteases [37]. Thus, the inhibition of microglia activation is able to reduce optic glioma proliferation in NF1 patients and influences the clinical variability of NF1 phenotype.
In addition to genotypes, functional phenotypic assays which integrate the different pathways provide useful tools to explore the role of DNA repair in inter-individual variability clinic. Methodologies for measuring DNA damage differ between laboratories and depend upon the DNA-damaging agent used, DNA repair kinetics, the endpoint measured and ways to measure the endpoint (quantitatively or qualitatively). In this sense, the alkaline comet assay protocol used in this study was adequate to detect significant differences in single strand breaks between NF1 patients and controls.
The alkaline comet assay tested was designed to provide the most comprehensive picture of the DNA damage induced, quantifying the cellular capacity to repair the observed lesions by showing the disappearance of damaged sites and the genome restoration. The assay was used to assess oxidative, baseline, H2O2-induced DNA damage and repair capacity of DNA and its related genetic instability in NF1 patients’ peripheral blood lymphocytes. H2O2 is a well-established genotoxic factor that can be used to evaluate the efficiency of DNA repair pathways as well as being used to assess resistance of cells to oxidant challenge. Exposure to hydrogen peroxide may result in DNA base damage and/or single- and double-strand breaks (SSBs and DSBs, respectively) due to the direct action or generation of free radicals [20]. Base modification and SSBs are repaired primarily by base excision repair (BER) [21, 38]. The majority of DSBs are repaired by nonhomologous end-joining (NHEJ) and homologous recombination repair (HRR) [39].
The use of peripheral blood lymphocytes was used based on the assumption that the DNA repair capacity of an individual is a genetic predisposition measurable in various cell types. Furthermore, this cellular population is easy to acquire from a blood draw and its measurements can serve as surrogates for other target tissues. This notion is supported by the results of studies on relatives and twins showing heritable repair phenotypes [40, 41].
Our main result was that the peripheral blood lymphocytes from case patients with NF1 showed decreased repair of damaged DNA than those from control subjects. We did not observe any difference between media baseline and oxidative endogenous level of DNA damage in lymphocytes of NF1 patients and subjects controls. These findings were consistent with other studies showing similar levels of constitutive DNA damage in the form of spontaneous chromosomal aberrations and sister chromatid exchanges (SCEs) in neurofibroma-derived cells and in normal skin fibroblasts, melanocytes, and peripheral blood lymphocytes among NF1 patients and controls [16, 17].
To the best of our knowledge, this is the first report on the assessment of endogenous damage, oxidized, induced, and unrepaired DNA damage in NF1 patients with the use of the comet assay. Further studies of genetic linkage and association are underway to identify the specific genetic variants associated with variable expression in NF1. Understanding the genetic mechanisms that control phenotypic expression in NF1 will provide further insight into the fundamental disease processes. All these raise the possibility that repair gene(s) playing a role in the pathogenesis of NF1 might be directly or indirectly implicated in pathways contributing to the control of genomic integrity.
Additionally, our results suggest that DNA repair kinetics measured by the comet assay may serve to identify the presence of genetic modifiers and would offer clues to the molecular pathogenesis of NF1. This hypothesis requires verification by long-term monitoring of the study patients and by correlates between DNA repair capacity and disease progression or severity. An altered expression of non-linked repair genes may eventually support more precise predictions of specific clinical features and complications of NF1 that could possibly lead to new therapeutic approaches.
In summary, no differences were found in the endogenous, oxidative and induced DNA damage by H2O2 between NF1 patients and healthy controls by using the comet assay. Nevertheless, there was a significant difference in repair kinetics in leukocytes of NF1 patients compared to the control group. Moreover, knowing that most of the patients involved in this study were children and that the frequency of more serious complications tends to increase with age, it would be important to carry on intensive follow-up and screening to all the patients, to see if they develop severe clinical events or not and to determine its relationship to their DNA repair capacity.
ACKNOWLEDGEMENTS
We want to thank Professor Iris Benzie from the Hong Kong Polytechnic University for review and criticism of the manuscript. We are grateful to the Cuban Ministry of Public Health for their financial support. We thank all the clinicians, patients and their families for their help.
REFERENCES
1. Friedman JM. Epidemiology of neurofibromatosis type 1. Am J Med Genet. 1999;89(1):1-6.
2. Cawthon RM, Weiss R, Xu GF, Viskochil D, Culver M, Stevens J, et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell. 1990;62(1):193-201.
3. Wallace MR, Marchuk DA, Andersen LB, Letcher R, Odeh HM, Saulino AM, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249(4965):181-6.
4. Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-8.
5. Upadhyaya M, Spurlock G, Monem B, Thomas N, Friedrich RE, Kluwe L, et al. Germline and somatic NF1 gene mutations in plexiform neurofibromas. Hum Mutat. 2008;29(8):E103-11.
6. Easton DF, Ponder MA, Huson SM, Ponder BA. An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): evidence for modifying genes. Am J Hum Genet. 1993;53(2):305-13.
7. Szudek J, Joe H, Friedman JM. Analysis of intrafamilial phenotypic variation in neurofibromatosis 1 (NF1). Genet Epidemiol. 2002;23(2):150-64.
8. Sabbagh A, Pasmant E, Laurendeau I, Parfait B, Barbarot S, Guillot B, et al. Unravelling the genetic basis of variable clinical expression in neurofibromatosis 1. Hum Mol Genet. 2009;18(15):2768-78.
9. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071-8.
10. Altieri F, Grillo C, Maceroni M, Chichiarelli S. DNA damage and repair: from molecular mechanisms to health implications. Antioxid Redox Signal. 2008;10(5):891-937.
11. Hazra TK, Das A, Das S, Choudhury S, Kow YW, Roy R. Oxidative DNA damage repair in mammalian cells: a new perspective. DNA Repair (Amst). 2007;6(4):470-80.
12. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17(10):1195-214.
13. Paz-Elizur T, Krupsky M, Elinger D, Schechtman E, Livneh Z. Repair of the oxidative DNA damage 8-oxoguanine as a biomarker for lung cancer risk. Cancer Biomark. 2005;1(2-3):201-5.
14. Paz-Elizur T, Elinger D, Leitner-Dagan Y, Blumenstein S, Krupsky M, Berrebi A, et al. Development of an enzymatic DNA repair assay for molecular epidemiology studies: distribution of OGG activity in healthy individuals. DNA Repair (Amst). 2007;6(1):45-60.
15. Tice RR, Agurell E, Anderson D, Burlinson B, Hartmann A, Kobayashi H, et al. Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen. 2000;35(3):206-21.
16. Schwenn MR, Weichselbaum RR, Little JB. Investigation of the cytotoxic effects of DNA damaging agents on neurofibromatosis cells. Mutat Res. 1985;142(1-2):55-8.
17. Troilo P, Strong LC, Little JB, Nichols WW. Spontaneous and induced levels of chromosomal aberration and sister-chromatid exchange in neurofibromatosis: no evidence of chromosomal hypersensitivity. Mutat Res. 1992;283(4):237-42.
18. Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175(1):184-91.
19. Nadin SB, Vargas-Roig LM, Ciocca DR. A silver staining method for single-cell gel assay. J Histochem Cytochem. 2001;49(9):1183-6.
20. Collins AR. Measuring oxidative damage to DNA and its repair with the comet assay. Biochim Biophys Acta. 2014;1840(2):794-800.
21. Collins AR, Gaivao I. DNA base excision repair as a biomarker in molecular epidemiology studies. Mol Aspects Med. 2007;28(3-4):307-22.
22. WMA Declaration of Helsinki - Ethical Principles for Medical Research Involving Human Subjects [Internet]. Ferney-Voltaire: World Medical Association, Inc.; 2013 [cited 2013 Oct 17]. Available from: http://www.wma.net/en/30publications/10policies/b3/.
23. Smith CC, O'Donovan MR, Martin EA. hOGG1 recognizes oxidative damage using the comet assay with greater specificity than FPG or ENDOIII. Mutagenesis. 2006;21(3):185-90.
24. Forchhammer L, Brauner EV, Folkmann JK, Danielsen PH, Nielsen C, Jensen A, et al. Variation in assessment of oxidatively damaged DNA in mononuclear blood cells by the comet assay with visual scoring. Mutagenesis. 2008;23(3):223-31.
25. Orlow I, Park BJ, Mujumdar U, Patel H, Siu-Lau P, Clas BA, et al. DNA damage and repair capacity in patients with lung cancer: prediction of multiple primary tumors. J Clin Oncol. 2008;26(21):3560-6.
26. Wiest V, Eisenbarth I, Schmegner C, Krone W, Assum G. Somatic NF1 mutation spectra in a family with neurofibromatosis type 1: toward a theory of genetic modifiers. Hum Mutat. 2003;22(6):423-7.
27. Wang Q, Montmain G, Ruano E, Upadhyaya M, Dudley S, Liskay RM, et al. Neurofibromatosis type 1 gene as a mutational target in a mismatch repair-deficient cell type. Human genetics. 2003;112(2):117-23.
28. Alotaibi H, Ricciardone MD, Ozturk M. Homozygosity at variant MLH1 can lead to secondary mutation in NF1, neurofibromatosis type I and early onset leukemia. Mutat Res. 2008;637(1-2):209-14.
29. Berwick M, Vineis P. Markers of DNA repair and susceptibility to cancer in humans: an epidemiologic review. J Natl Cancer Inst. 2000;92(11):874-97.
30. Kruger S, Kinzel M, Walldorf C, Gottschling S, Bier A, Tinschert S, et al. Homozygous PMS2 germline mutations in two families with early-onset haematological malignancy, brain tumours, HNPCC-associated tumours, and signs of neurofibromatosis type 1. Eur J Hum Genet. 2008;16(1):62-72.
31. Sabbagh A, Pasmant E, Laurendeau I, Parfait B, Barbarot S, Guillot B, et al. Unravelling the genetic basis of variable clinical expression in neurofibromatosis 1. Hum Mol Genet. 2009;18(15):2768-78.
32. Gutmann DH, Winkeler E, Kabbarah O, Hedrick N, Dudley S, Goodfellow PJ, et al. Mlh1 deficiency accelerates myeloid leukemogenesis in neurofibromatosis 1 (Nf1) heterozygous mice. Oncogene. 2003;22(29):4581-5.
33. Paunu N, Syrjakoski K, Sankila R, Simola KO, Helen P, Niemela M, et al. Analysis of p53 tumor suppressor gene in families with multiple glioma patients. J Neurooncol. 2001;55(3):159-65.
34. Elmariah SB, Huse J, Mason B, Leroux P, Lustig RA. Multicentric glioblastoma multiforme in a patient with BRCA-1 invasive breast cancer. Breast J. 2006;12(5):470-4.
35. Kruger S, Kinzel M, Walldorf C, Gottschling S, Bier A, Tinschert S, et al. Homozygous PMS2 germline mutations in two families with early-onset haematological malignancy, brain tumours, HNPCC-associated tumours, and signs of neurofibromatosis type 1. Eur J Hum Genet. 2008;16(1):62-72.
36. El-Zein R, Bondy ML, Wang LE, de Andrade M, Sigurdson AJ, Bruner JM, et al. Increased chromosomal instability in peripheral lymphocytes and risk of human gliomas. Carcinogenesis. 1999;20(5):811-5.
37. Rosado MM, Bennici E, Novelli F, Pioli C. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology. 2013;139(4):428-37.
38. El-Zein RA, Monroy CM, Cortes A, Spitz MR, Greisinger A, Etzel CJ. Rapid method for determination of DNA repair capacity in human peripheral blood lymphocytes amongst smokers. BMC Cancer. 2010;10:439.
39. Parshad R, Sanford KK. Radiation-induced chromatid breaks and deficient DNA repair in cancer predisposition. Crit Rev Oncol Hematol. 2001;37(2):87-96.
40. Surowy H, Rinckleb A, Luedeke M, Stuber M, Wecker A, Varga D, et al. Heritability of baseline and induced micronucleus frequencies. Mutagenesis. 2011;26(1):111-7.
41. Lin J, Swan GE, Shields PG, Benowitz NL, Gu J, Amos CI, et al. Mutagen sensitivity and genetic variants in nucleotide excision repair pathway: genotype-phenotype correlation. Cancer Epidemiol Biomarkers Prev. 2007;16(10):2065-71.
Received in August, 2013.
Accepted in April, 2014.
Reinaldo Gutierrez. Centro Nacional de Genética Médica, CNGM. Calle 146 No. 3102. Playa, CP 10600, La Habana, Cuba. E-mail: rey@infomed.sld.cu.

{kind=link}