










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBiotecnología Aplicada
versión On-line ISSN 1027-2852
Biotecnol Apl vol.33 no.2 La Habana abr.-jun. 2016
REVIEW
Oxidative stress in pharmacoresistant epilepsy
Estrés oxidativo en la epilepsia resistente a fármacos
Lourdes Lorigados1, Lilia M Morales2, Sandra Orozco-Suárez3, Juan M Gallardo4, Mei L Díaz-Hung1, María E González1, Bárbara Estupiñán5, Nancy Pavón1, Luisa Rocha6
1 Departamento de Inmunoquímica, Centro Internacional de Restauración Neurológica, Ciren. Ave. 25 No. 15805 e/ 158 y 160, CP 11300, Playa, La Habana, Cuba.
2 Servicio de Neurofisiología Clínica, Ciren, La Habana, Cuba.
3 Unidad de Investigación Médica en Enfermedades Neurológicas, Hospital de Especialidades, Centro Médico Nacional Siglo XXI, Instituto Mexicano del Seguro Social, México.
4 Unidad de Investigación Médica en Enfermedades Nefrológicas, Hospital de Especialidades. Centro Médico Nacional Siglo XXI, Instituto Mexicano del Seguro Social, México.
5 Laboratorio de Morfología, Ciren, La Habana, Cuba.
6 Departamento de Farmacobiología, CINVESTAV, Sede Sur, DF, México.
ABSTRACT
Pharmacoresistant epilepsy is a phenomenon increasingly affecting a substantial proportion of patients treated with current anti-epileptic drugs, who became refractive to therapy. Coincidently, anti-epileptic drugs have been related to oxidative stress (OS)-related processes, which could have an impact in further drug development and also disease progression. Therefore, this review is aimed to analyzing the OS processes resulting in excitotoxicity, neuroinflammation or mitochondrial dysfunction, which have been implicated in numerous neurological disorders, and particularly its role in epilepsy. Evidence from clinical data and a variety of animal models of temporal lobe epilepsy (TLE) is discussed, regarding damage to proteins, lipids, and antioxidant defenses. An emerging overall picture on the relationship of OS with cell and soluble mediators of inflammation and excitotoxicity is presented. Moreover, new therapeutic strategies are discussed, as proposed to complement current therapies or to develop new ones which could effectively interfere with the chronic changes induced by recurrent seizures for a better control on the progression of the disease.
Keywords: epilepsy, pharmacoresistance, oxidative stress, neuroinflammation, antiepileptic drug therapies.
RESUMEN
La epilepsia resistente a fármacos es una condición que afecta a una proporción sustancial de pacientes tratados con los fármacos antiepilépticos disponibles, los que devienen refractarios a la terapia. Coincidentemente, los fármacos antiepilépticos han sido relacionados con procesos de estrés oxidativo (OS), lo cual puede impactar en la progresión de la enfermedad y ser relevante para el sucesivo desarrollo de fármacos. En esta revisión se analizan los procesos de OS vinculados a la epilepsia resistente a fármacos, que derivan en procesos de excitotoxicidad, neuroinflamación o disfunción mitocondrial, que se han manifestado en otros desórdenes neurológicos y particularmente en la epilepsia. Se muestran evidencias basadas en datos obtenidos mediante el estudio de la epilepsia del lóbulo temporal (ELT) en modelos animales, que derivan en daño a las proteínas, los lípidos y las defensas antioxidantes. Se incluye una vista general emergente sobre la relación del OS y los mediadores solubles involucrados de la inflamación y la excitotoxicidad. Además, se discuten nuevos abordajes terapéuticos, como los propuestos para complementar las terapias existentes o incluso nuevas estrategias para interferir los cambios crónicos inducidos por los ataques recurrentes para un mejor control de la progresión de la enfermedad.
Palabras clave: epilepsia resistente a fármacos, estrés oxidativo, neuroinflamación, terapias con fármacos antiepilépticos.
INTRODUCTION
Epilepsy is one of the most common and serious brain disorders. It affects nearly 50 million people worldwide. It is characterized by recurrent spontaneous seizures due to an imbalance between cerebral excitability and inhibition [1]. This leads to uncontrolled excitability, which generates an imbalance in the metabolic rate of the brain. Nevertheless, the molecular mechanisms that lead to seizures and epilepsy are not well understood.
The incidence of epilepsy is about 2 % and approximately 60-80 % of patients can be controlled with antiepileptic drugs [2]. In more than 60 % of cases, seizures remit permanently. Nevertheless, a substantial proportion of patients (30 %) do not respond to antiepileptic drugs (AEDs) medication, despite its administration in an optimally monitored regimen. Such cases are often loosely termed intractable or pharmacoresistant epilepsy [3]. Most of them suffer from a focal form of epilepsy. The areas of epileptogenesis are usually characterized by cell loss [4, 5]. Simultaneously, AEDs could also contribute to the established brain damage through their diverse effects on the antioxidative system [6, 7], some of them potentially generating reactive oxygen species (ROS) and triggering oxygen-dependent tissue injury [8]. Whereas a precise role for ROS in the epilepsies remains to be defined, a general role for ROS in seizure-induced neuronal death is supported in part by the observations that repeated seizures result in increased oxidation of cellular macromolecules [9]. Moreover, there is an intrinsic relationship between oxidative stress (OS) and inflammation in epileptic patients, as a source of neuronal decline, manifested in a series of pathological etiologies and particularly in pharmacorresistant epilepsy.
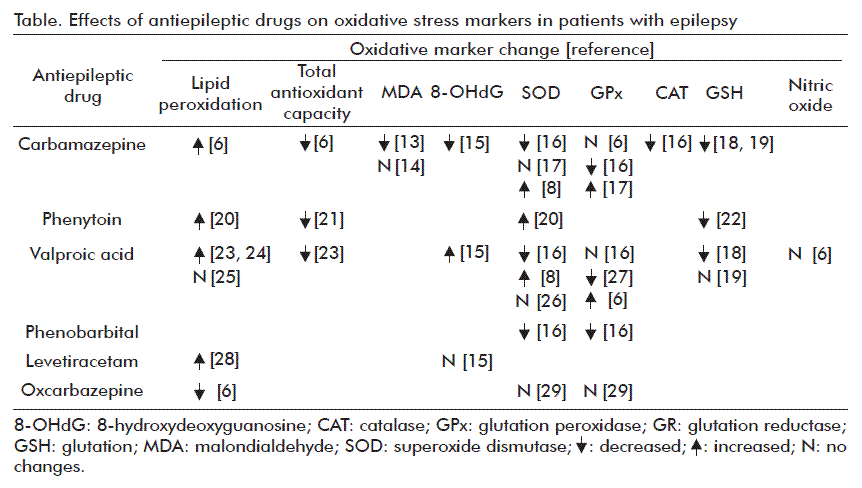
The use of AEDs with possible neuroprotective effects has been investigated in human or animal models [10, 11]. Furthermore, AEDs pro-oxidative effects might lead to the enhancement of seizure activity, which can result in loss of AEDs efficacy or apparent functional tolerance, pharmacotherapy resistance and undesired side effects. Studies have been performed in serum, plasma, erythrocytes or leukocytes [12], some of their effects on OS markers in epileptic patients summarized in the table.
During metabolic processes, numerous AEDs as phenobarbitone (PB), phenytoin (PHT), carbamazepine (CBZ) and valproic acid (VPA), produce reactive metabolites which can increase the formation of ROS that can induce oxidative damage and cause toxicity. To support this theory, numerous studies evaluating the influence of epilepsy and AEDs on the formation of free radicals, show that either of them could be connected to oxidative stress generation [30]. Moreover, AEDs may also provoke systemic toxicity, in addition to their main inhibitory activity on the epileptic focuses, either through increased oxidative damage or covalent binding of their reactive metabolites to biological macromolecules [23, 31].
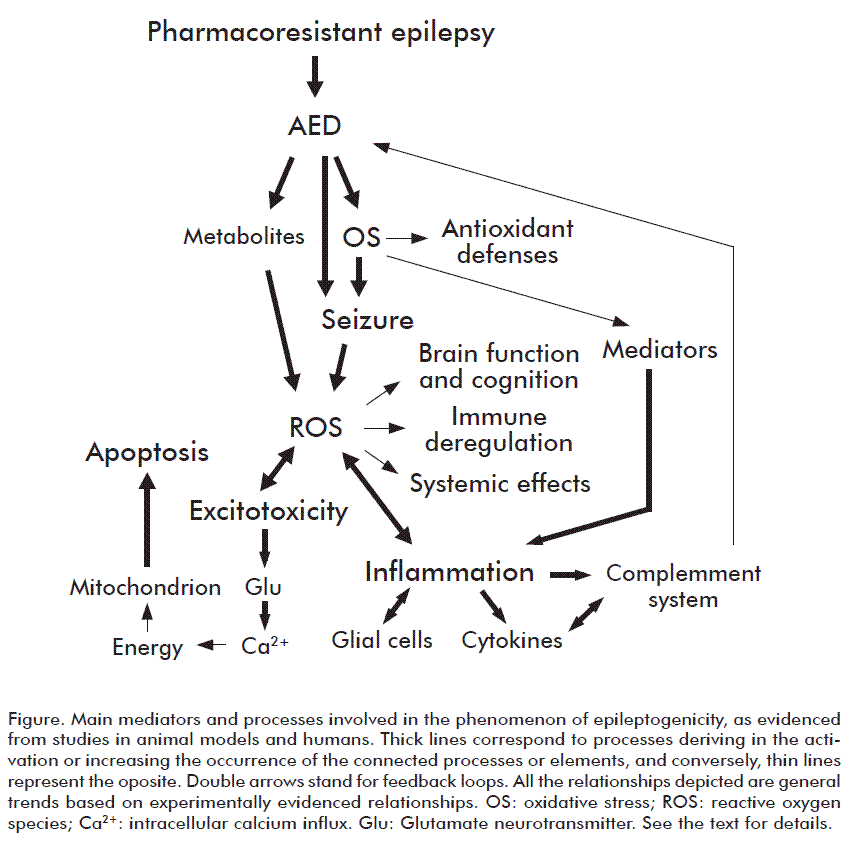
Therefore, in this review, we address the picture arising from the connection among pharmacoresistant epilepsy and OS, and their relation to inflammation (Figure). Results in experimental models and in patients that support the presence of an imbalance in the oxidative status in pharmacoresistant epilepsy are also discussed, with emphasis on pharmacoresistant temporal lobe epilepsy (TLE).
PARTICIPATION OF OXIDATIVE STRESS IN EPILEPSY
Oxidative stress (OS) is a biochemical state in which reactive oxygen species (ROS) are generated, been involved in diverse physiological and pathological conditions, including epilepsy. At high concentrations, ROS react readily with proteins, lipids, carbohydrates, and nucleic acids, often inducing signaling and redox control disruption, and irreversible functional alterations of molecules or even their complete destruction [32].
ROS have a crucial role in human physiological and pathophysiological processes [33]. They were originally considered to be exclusively detrimental to cells [34], but recent evidence suggests that redox regulation involving ROS is essential for the modulation of critical cellular functions such as mitogen-activated protein (MAP) kinase cascade activation, ion transport, calcium mobilization, apoptosis program activation, and other signaling mechanisms. At the systemic level they contribute to complex functions such as blood pressure regulation, cognitive function and immune function. ROS enable the response to growth factor stimulation and the generation of the inflammatory response [33]. Therefore, OS is involved also in acute and chronic CNS through the action of free radicals-mediated injury and is a major factor in the pathogenesis of neuronal damage [35].
The brain is believed to be particularly vulnerable to OS as it contains high concentrations of polyunsaturated fatty acids that are susceptible to lipid peroxidation, consumes relatively large amounts of oxygen for energy production, and has lower antioxidant defenses compared to other organs, making OS a likely contributor to neurological disorders [12, 35, 36]. The central nervous system (CNS) has an extraordinary metabolic rate consuming approximately 20 % of all inhaled oxygen at rest; however, it only accounts for 2 % of body weight. This enormous metabolic demand is due to the fact that neurons are highly differentiated cells and need large amounts of ATP in order to maintain ionic gradients across cell membranes and for neurotransmission [37].
Recently, accumulating evidence supports the association between OS and seizures, in the process of their generation, and in the mechanisms associated with refractoriness to drug therapy. Alterations in the antioxidant enzymes [38-40] and increases in the indicators of oxidative damage to biomolecules, such as malondialdehyde (MDA), protein carbonyls and 8-hydroxy-2-deoxyguanosine and activation of the nicotinamide adenine dinucleotide phosphate oxidase have been reported [39, 41]. Similarly, data from animal studies suggest that prolonged seizure activity might result in the increased production of ROS and generation of nitric oxide and peroxynitrite preceded neuronal cell death in vulnerable brain regions [42, 43].
Previous studies have demonstrated that seizure-induced mitochondrial dysfunction and excess free radical production cause oxidative damage to cellular components and initiate the mitochondrial apoptotic pathway [44, 45]. OS is also considered an important consequence of excitotoxicity and inflammation, two of the proposed mechanisms for seizure-induced brain damage [45-48] and finally, there are evidences of the changes on oxidative markers when some AEDs are consumed [12].
OXIDATIVE STRESS AND INFLAMMATION IN EPILEPSY
Inflammation, in turn, appears to play a central role among the various mechanisms that have been connected to epileptogenic process [49-53]. The oxidative and nitrosative stress pathways are induced by inflammatory responses, and subsequent mitochondrial metabolic processes generate highly reactive free radical. There are different pathways of ROS generation, and one of them is through cellular pathway like cytokines receptors [54].
Several studies have suggested a link between OS and inflammation [55, 56]. Some authors have implicated OS as a major upstream component in the signaling cascade involved in activation of redox-sensitive transcription factors and proinflammatory gene expression leading to inflammatory response [57].
Although often overlooked, glial cells, such as astrocytes and microglia, play important roles in maintaining overall CNS homeostasis, providing trophic support to neurons, clearing synapses of the released neurotransmitters, mediating immune responses in the brain, and reducing OS [58]. When CNS regional homeostasis is disturbed because of redox shifts or other neurodegenerative conditions, astrocytes and microglia release various cytokines in an effort to re-establish regional integrity and repair damaged cells. While these glial responses are beneficial to neurons, the continuous or repeated activation of astrocytes and microglia under conditions of chronic inflammatory stresses can lead to the increased production of ROS/RNS, which can lead to severe neuronal damage [59, 60].
In special, evidence of brain inflammation has been found to be associated with diverse pathological etiologies in patients with epilepsy and in special in pharmacorresistant epilepsy. For example, proinflammatory molecules, reactive astrocytosis, activated microglia, and other indicators of inflammation have been found in the hippocampi of patients with TLE, in and around epileptic tubers in patients with tuberous sclerosis, and in some epileptic cases with cortical dysplastic lesions [46, 61-64].
Data collected using tissue of patients with TLE suggests that specific inflammatory pathways are chronically activated during epileptogenesis and that they persist in chronic epileptic tissue, contributing to the etiopathogenesis of TLE [62]. Hippocampus obtained from patients with hippocampal sclerosis (HS) shows microglial activation [61, 62]. Expression of proinflammatory molecules (IL-1, IL-6 and TNF) as well as IL-1α, IL-1β and IL-1 receptor type I, NFkB. Also the complement system is augmented in epileptic tissue surgically removed from patients with pharmacoresistant epilepsy [65]. Systemic IL-6 levels in peripheral blood are increased immediately after seizures and long lasting during post-ictal period (24 h after ictal event) in patients with TLE, an effect not detected in patients with HS [66]. Expression of C1q, C3c, and C3d is augmented within regions where neuronal cell loss occurs. Other studies indicate the activation of complement pathway, involving both reactive astrocytes and cells of the microglia/macrophage lineage in human HS specimens [61]. These observations suggest the existence of a feedback loop between the pro-inflammatory cytokine system and components of the complement cascade, which may be critical for the propagation of the inflammatory response in human TLE with HS and therefore consequent OS as a result of the inflammation.
Thus, the mechanisms related to OS and inflammation in epileptic patients must be further explored in the clinical management of these conditions.
OXIDATIVE STRESS AND EXCITOTOXICITY IN EPILEPSY
OS is thought to be an important consequence of glutamate receptor activation and excitotoxicity which plays a critical role in epileptic brain damage. There is evidence supporting the hypothesis that the neurodegenerative changes associated with human epilepsy arise from persistent discharges in the glutamate pathway [36]. The mechanism is relatively simple: excess of glutamate release leads to repeated depolarization-repolarization cycles in glutamate terminals, until glutamate reaches toxic concentrations and, finally, the excitotoxic degeneration of post-synaptic neurons takes place [67].
In general, excitotoxic damage to the neurons of epileptic patients is mediated by excessive calcium inflow during seizures. The excessive elevation of the cytoplasmic Ca2+ concentration could promote: 1. the synthesis of nitric oxide, 2. the generation of free radicals, as superoxide or peroxynitrites, and 3. the loss of electrochemical mitochondrial potential, altering the oxidative phosphorylation and promoting the free radicals generation until complete invalidation of the mitochondrial metabolism, which could lead to the ending of the energy cellular reserves. Furthermore, the rising in Ca2+ cytoplasmic concentration activatesvarious intracellular signaling pathways dependent on protein kinases and phosphatases that could promote proteolysis of the cellular content [47, 68, 69]. Finally, procaspases are activated likewise, and neuronal death eventually takes place by necrosis, apoptosis or autophagy [48].
Taken these evidences together, it is now clear that excitatory amino acid and ROS may cooperate in the pathogenesis of neuronal damage, involving loss of cellular calcium homeostasis. Excitatory events may stimulate ROS, and there is evidence that ROS can lead to release of excitatory amino acid, suggesting a bidirectional relationship.
EXPERIMENTAL AND CLINICAL EVIDENCES OF OXIDATIVE STRESS AND MITOCHONDRIAL DYSFUNCTION IN PHARMACORESISTANT EPILEPSY
Experimental models
The use of animal models has made important contributions to our understanding of seizures. The most popular models of excitotoxicity employing adult animals are those based on the use of kainic acid (KA) and pilocarpine. These are models of TLE, induced by the unilateral or systemic injection of these compounds at convulsant doses, causing excitotoxic damage at the
pyramidal neurons of hippocampus and the hilar region. The models based on systemically administering KA or pilocarpine are widely used for studying generalized tonic-clonic convulsions or the epileptic state, whose neuroanatomical substrate is temporal mesial sclerosis [70]. Liang et al. have demonstrated that 16 hours after KA injection, the enzyme aconitase becomes inactive decreasing the availability of reducing agents for the mitochondrial electron transport chain and compromising ATP synthesis [71].
During other experiment in which KA was injected directly into the CA3 area of the hippocampus, an increase in nitric oxide synthesis was demonstrated, contributing to cell death by apoptosis in the CA3 area of the hippocampus after the induction of a status epilepticus [42]. Therefore in the KA induction model there is an increase in ROS production, mitochondrial dysfunction, and apoptosis of neurons in several areas of the brain, especially those in the hippocampus [72].
Other recent research have shown an acute increase in mitochondrial hydrogen peroxide production, an index of mitochondrial OS, and oxidative damage to mitochondrial DNA up to 96 h following KA and lithium pilocarpine induced epileptogenesis [73-75]. Lipid peroxidation is a pathway suggested as a mechanism of epileptic activity [76]. Whereas the MDA has been used to identify oxidative damage to lipids acutely following seizure events [77] and their levels have been reported to be increased up to 16 h following KA treatment in the hippocampus [78], up to 24 h in an amygdala kindling model of epilepsy [79], and 2 h post-pilocarpine-induced status epilepticus in the cortex [80].
There is evidence to support an increase in ROS production in epilepticus induced by pilocarpine or KA, producing considerable amounts of superoxide and overloading endogenous protection mechanisms (GPx, proteins, phospholipids, and mitochondrial DNA [81]. Other authors reported hydroperoxide to be increased at 1 h post-pilocarpine treatment [82]. A large increase in stable arachidonic acid derived prostaglandin products of lipid oxidation, including F2-isoprostanes and isofurans has been shown early after status epilepticus in hippocampal subregions [83].
On the other hand, Kudin et al. [84] have shown a mitochondrial dysfunction several weeks after pilocarpine-induced status epilepticus. The activities of complexes I and IV of the electron transport chain decrease and complex II increases 1 month after pilocarpine status epilepticus. They found also lowered mitochondrial membrane potential in the CA1 and CA3 areas. These changes may be attributed to decreased mitochondrial DNA copy number that results in down regulation of oxidative phosphorylation enzymes encoded by mitochondrial DNA [84].
Recently, Waldbaum et al. investigated whether acute lesions induced by ROS formation contribute to the formation of chronic epilepsy [85]. They have questioned whether mitochondrial and cellular alterations might occur during the ‘latency period’ between the initial brain lesion and the appearance of recurring spontaneous seizures, inducing progression to chronic epilepsy. An adaptive increase of mitochondrial DNA repair occurs immediately after ROS increase induced by acute status epilepticus. However, chronic increase in ROS production is accompanied by failure in the induction of mitochondrial DNA repair [73]. OS markers as glutation and specific markers of redox status in the mitochondrion (coenzyme A) have recently been demonstrated to decrease in the hippocampus after lithium pilocarpine induced status epilepticus and to become permanently damaged during epileptogenesis and chronic epilepsy, even when hydrogen peroxide production measurements and mitochondrial DNA damage return to control levels [74]. This may contribute to significant mitochondrial dysfunction, harming neuronal excitability through electron transport chain dysfunction and decreased ATP production.
In summary, status epilepticus and other epileptogenic injuries result in mitochondrial dysfunction, increased ROS formation and oxidative damage to proteins, lipids and DNA in different experimental models of epilepsy.
Clinical evidences
Evidences of mitochondrial dysfunction and OS during chronic epilepsy have been recently found in TLE patients [86, 87]. For example, Kunz et al. have been finding mitochondrial complex I deficiency in the seizure foci [88].
Previously studies found increased activities of SOD, CAT, markers of lipid peroxidation and decreased activities of GPx in pharmacoresistant TLE patients [24, 89]. Sudha et al. reported in 2001 decreased glutathione reductase activity. The lipid peroxidation and percentage haemolysis were higher compared to controls. Furthermore, erythrocyte glutathione reductase and plasma ascorbate and vitamin A concentrations were lower [86]. Meanwhile, many different studies were noted increased markers of lipid peroxidation [39, 89, 90].
On the other hand, there are also other studies have not detected changes in SOD, CAT, GPx and glutation reductase activities [86, 89, 91, 92].
Our group has been investigating the impact of the epilepsy surgery on serum markers of oxidative damage in pharmacoresistant TLE patients [39]. Before surgery, we found increased activities of SOD, CAT, markers of lipid peroxidation and decreased activities of GPx. An interesting finding was the positive correlation between duration of the disease and advanced oxidation protein product levels. This result suggests the early presence of oxidative damage to proteins in initial stages of the illness. This could be due to protein repairing mechanisms that do not act as efficiently as in other biomolecules.
After surgery, the patients showed a tendency to normalization of the studied variables, except for SOD activity. The outlying redox state of the patients markedly improved after surgery, which is clearly evidenced by an important decrease in MDA and advanced oxidative protein products levels two years after surgery. The recovery in GPx activity was also notorious, as it contributes to a decrease in oxidative damage and a better redox balance [39]. On the other hand, we can speculate that the sustained increase in superoxide dismutase activity could recede if the epileptoid activity in the remaining regions eventually disappears in these patients. Finally, the increase in CAT activity levels seems to be a cellular response to the intense ROS production triggered by seizure episodes.
Other recent results from our group showed alterations in oxidative markers in tissue from 16 TLE pharmacoresistant patients who received surgical treatment. There are increases of lipid peroxidation measured in terms of tissue MDA and altered antixodative defenses (SOD, GPx, unpublished data).
When we evaluated the cellular immunity (CD8+, CD25+ and HLA-DR+ cells) and inflammatory markers (IL-1β, IL-6) in the same patients before surgical treatment, both parameters were found increased. One year after surgical treatment, we have detected a decrease in all markers (cellular immunity and inflammatory response) when compared with presurgical values [46]. A previous study of brain tissue from the same patients evidenced an increase in immunopositivity to Anexin-V [93] and this correlated positively with the IL-6, and IL-1β [94]. This finding supports a relationship between the inflammatory process in epilepsy and the neuronal death observed in neocortical tissue from these patients. These results indicate that once the epileptogenic zone is resected and seizure activity is decreased, there is a reduction in proinflammatory cytokines suggesting that seizures are the cause of the inflammatory disorders observed in patients with drug-resistant epilepsy [46, 94]. It is of particular interest to note that one year after surgery 75 % of these patients were free of seizures and the rest of the patients showed a significant decrease in the number of episodes [95].
In summary, our results indicate altered antioxidative defenses and damage to biomolecules, immunological and inflammatory alteration in patients with pharmacoresistant TLE [39]. Further studies are needed to ascertain whether ROS are involved in the pathogenesis of pharmacoresistant TLE and the association with inflammatory mechanisms that take place in the pathophysiology of this type of epilepsy. Considering this knowledge would be interesting to correlate the oxidative markers in TLE pharmacoresistant patients and levels of proinflammatory mediators in order elucidate the relationship between oxidative imbalance described for these patients and these mechanisms.
THERAPEUTIC POTENTIAL FOR PHARMACORESISTANT EPILEPSY
Therapies supplementary to AEDs have been proposed to increase their effectiveness and their testing is underway, most of them counteracting the action of ROS through the enhancement of antioxidant defenses or by providing antioxidant-rich supplementary diets.
Such strategies are based on the distribution of protective antioxidants in the body, which shows some interesting features: there is a relatively high concentration of the water-soluble antioxidant vitamin C in the brain, while vitamin E concentrations in CNS are not remarkably different from those in other organs. The concentrations of antioxidants also vary within the different regions of the brain itself. For instance, the lowest concentration of vitamin E is found in the cerebellum. It was also shown that enzymatic antioxidants, such as CAT, are at lower concentrations in the brain than in other tissues [96].
Antioxidants are exogenous (natural or synthetic) or endogenous compounds acting in several ways including removal of superoxide, scavenging ROS or their precursors, inhibiting ROS formation and binding metal ions needed for catalysis of ROS generation [97]. Among those showing enzymatic activity, CAT, SOD, GPx), glutathione reductase and thioredoxin exhibit biological value [98]. Particularly, SOD has been shown to protect against programmed cell death [99]. The non-enzymatic antioxidants are actually the scavengers of ROS and reactive nitrogen species (RNS). These include glutathione, vitamins E and C (inhibits oxidation of membrane lipid). Uric acid is the scavenger of peroxynitrite in plasma, albumin, bilirubin, N-Acetylcysteine, melatonin which directly reacts with ROS and form disulfides.
Other antioxidant compounds such as lipoic acid, ascorbic acid and alphatocopherol can protect the brain against OS [100-102]. In animal models of TLE, the OS and neuronal damage, but not behavioral seizures induced by kainate can be ameliorated by at least two types of SOD mimetics, the manganese porphyrin MnTBAP [71] and the compound EUK134 [103]. Other compounds with antioxidant properties that inhibit seizure-induced brain injury include the hormone melatonin. Melatonin stimulates gene expression for the antioxidant enzymes and increases their activity. Additionally, it neutralizes the hydroxyl, superoxide and peroxyl radicals, peroxynitrite anion, singlet oxygen, hydrogen peroxide, nitric oxide, and hypochlorous acid [104].
Kong et al. have investigated the role of RNA oxidation in epileptogenesis. Using pilocarpine to induce SE, they observed a significant increase in RNA oxidation in vulnerable neurons in rat brains immediately after SE followed by neuronal death [105]. However, a daily supplement of antioxidants (coenzyme Q10) significantly reduced RNA oxidation and protected rats from status epilepticus and neuronal loss [105].
In the case of Vitamin E (as α-tocopherol), it displays beneficial effects in epilepsy, mainly ascribed to its antioxidant properties. Ambrogini et al. showed that α-tocopherol oral supplementation before inducing status epilepticus, markedly reduces astrocytic and microglial activation, neuronal cell death and oxidative stress in the hippocampus from kainic acid-induced epilepsy rats [106]. However, clinical trials of vitamin E as an add-on therapy for refractory epilepsy have been controversial, with largely failed attempts to influence the occurrence of epileptic seizures in pediatric patients [9].
Additionally, Creatine supplementation has been effective in reducing hypoxia-induced seizures in animal models [107]. The finding that newer antiepileptic drugs such as zonisamide possess antioxidant properties raises the possibility that free radical scavenging may in part support their antiepileptic actions [108].
On other hand, ketogenic diets (i.e., high-fat, low carbohydrate diet) are used as another therapy for intractable epilepsy, although its mechanism of action is unknown. The proposed mechanism relies on the change of energy metabolism to utilization of fatty acids and their metabolites, ketonic bodies [109-111]. Recent works suggest that chronic consumption of a ketogenic diet may alter mitochondrial function by chronically decreasing production of ROS, increasing the expression of uncoupling proteins, promoting mitochondrial biogenesis, and stimulating GSH biosynthesis [112, 113]. Jarret et al. have demonstrated that the ketogenic diet specifically enhances the antioxidant capacity of brain mitochondria and showed evidence that GSH synthesis is up-regulated in ketogenic diet fed rats [112].
Studies have shown that this diet is as good as, or better than, any of the newer medications in reducing seizure frequency. However, concerns about adverse effects have been raised. Zamani et al. showed in an open label trial the effects of this diet on serum lipid profile. Results of this study indicate that a classic ketogenic diet in children with refractory seizures is effective in seizure reduction, but leads to development of hypercholesterolemia and hypertriglyceridemia [114].
Finally, development of therapies influencing mitochondrial energetic and OS for the epilepsies will ultimately depend on unraveling their role in the disease process.
CONCLUDING REMARKS
Clinical and preclinical data support the participation of OS and mitochondrial dysfunction in the epileptic process, suggesting that specific inflammatory pathways are chronically activated in the epileptogenic brain tissue. These results highlight the need for research that enables us to understand the role of the OS in the pathogenesis of pharmacoresistant epilepsy and particularly, to clarify the relationship between OS, inflammation and immunological deficit as physiopathological mechanism in TLE. OS and mitochondrial dysfunction occur as a consequence of prolonged epileptic seizures and influence seizure-induced brain injury. Conversely, OS can render the brain more susceptible to epileptic seizures. Therefore, OS and mitochondrial dysfunction may be both an important cause and a consequence of prolonged seizures.
Insight into the mechanisms by which seizures initiate OS and mitochondrial dysfunction and vice versa may provide novel therapeutic approaches for the treatment of epilepsies.
Further investigations into the role of inflammation and the immune response in CNS, particularly in pharmacoresistant epilepsy may add important insights in the understanding of the epileptogenic mechanism and open new ways of neuromodulatory treatment of epilepsy.
REFERENCES
1. World Health Organization. Epilepsy Fact Sheet. 2016 Feb [cited 2016 Feb 7]. Available from: http://www.who.int/mediacen-tre/factsheets/fs999/en/
2. Hauser WA, Annegers JF, Kurland LT. Prevalence of epilepsy in Rochester, Minnesota: 1940-1980. Epilepsia. 1991;32(4):429-45.
3. Regesta G, Tanganelli P. Clinical aspects and biological bases of drug-resistant epilepsies. Epilepsy Res. 1999;34(2-3):109-22.
4. Henshall DC, Meldrum BS. Cell death and survival mechanisms after single and repeated brief seizures. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. 4th ed. Bethesda (MD)2012.
5. Fujikawa DG, Shinmei SS, Cai B. Kainic acid-induced seizures produce necrotic, not apoptotic, neurons with internucleosomal DNA cleavage: implications for programmed cell death mechanisms. Neuroscience. 2000;98(1):41-53.
6. Hamed SA, Abdellah MM, El-Melegy N. Blood levels of trace elements, electrolytes, and oxidative stress/antioxidant systems in epileptic patients. J Pharmacol Sci. 2004;96(4):465-73.
7. Solowiej E, Sobaniec W. The effect of antiepileptic drug therapy on antioxidant enzyme activity and serum lipid peroxidation in young patients with epilepsy. Neurol Neurochir Pol. 2003;37(5):991-1003.
8. Karikas GA, Schulpis KH, Bartzeliotou A, Regoutas S, Thanopoulou C, Papaevangelou V, et al. Early effects of sodium valproate monotherapy on serum paraoxonase/arylesterase activities. Scand J Clin Lab Invest. 2009;69(1):31-5.
9. Patel M. Mitochondrial dysfunction and oxidative stress: cause and consequence of epileptic seizures. Free Radic Biol Med. 2004;37(12):1951-62.
10. Azam F, El-Gnidi BA, Alkskas IA. Combating oxidative stress in epilepsy: design, synthesis, quantum chemical studies and anticonvulsant evaluation of 1-(substituted benzylidene/ethylidene)-4-(naphthalen- 1-yl)semicarbazides. Eur J Med Chem. 2010;45(7):2817-26.
11. Azam F, Prasad MV, Thangavel N. Targeting oxidative stress component in the therapeutics of epilepsy. Curr Top Med Chem. 2012;12(9):994-1007.
12. Martinc B, Grabnar I, Vovk T. The role of reactive species in epileptogenesis and influence of antiepileptic drug therapy on oxidative stress. Curr Neuropharmacol. 2012;10(4):328-43.
13. Sobaniec W, Solowiej E, Kulak W, Bockowski L, Smigielska-Kuzia J, Artemowicz B. Evaluation of the influence of antiepileptic therapy on antioxidant enzyme activity and lipid peroxidation in erythrocytes of children with epilepsy. J Child Neurol 2006 Jul;21(7):558-62.
14. Safar MM, Abdallah DM, Arafa NM, Abdel-Aziz MT. Magnesium supplementation enhances the anticonvulsant potential of valproate in pentylenetetrazol-treated rats. Brain Res. 2010;1334:58-64.
15. Varoglu AO, Yildirim A, Aygul R, Gundogdu OL, Sahin YN. Effects of valproate, carbamazepine, and levetiracetam on the antioxidant and oxidant systems in epileptic patients and their clinical importance. Clin Neuropharmacol. 2010;33(3):155-7.
16. Niketic V, Ristic S, Saicic ZS, Spasic M, Buzadzic B, Stojkovic M. Activities of antioxidant enzymes and formation of the glutathione adduct of hemoglobin (Hb ASSG) in epileptic patients with long-term antiepileptic therapy. Farmaco. 1995;50(11):811-3.
17. Johannessen SI, Landmark CJ. Antiepileptic drug interactions - principles and clinical implications. Curr Neuropharmacol. 2010;8(3):254-67.
18. Cengiz M, Yuksel A, Seven M. The effects of carbamazepine and valproic acid on the erythrocyte glutathione, glutathione peroxidase, superoxide dismutase and serum lipid peroxidation in epileptic children. Pharmacol Res. 2000;41(4):423-5.
19. Ono H, Sakamoto A, Sakura N. Plasma total glutathione concentrations in epileptic patients taking anticonvulsants. Clin Chim Acta. 2000;298(1-2):135-43.
20. Liu CS, Wu HM, Kao SH, Wei YH. Serum trace elements, glutathione, copper/zinc superoxide dismutase, and lipid peroxidation in epileptic patients with phenytoin or carbamazepine monotherapy. Clin Neuropharmacol. 1998;21(1):62-4.
21. Mahle C, Dasgupta A. Decreased total antioxidant capacity and elevated lipid hydroperoxide concentrations in sera of epileptic patients receiving phenytoin. Life Sci. 1997;61(4):437-43.
22. Karabiber H, Yakinci C, Durmaz Y, Temel I, Mehmet N. Serum nitrite and nitrate levels in epileptic children using valproic acid or carbamazepine. Brain Dev. 2004;26(1):15-8.
23. Schulpis KH, Lazaropoulou C, Regoutas S, Karikas GA, Margeli A, Tsakiris S, et al. Valproic acid monotherapy induces DNA oxidative damage. Toxicology. 2006;217(2-3):228-32.
24. Yis U, Seckin E, Kurul SH, Kuralay F, Dirik E. Effects of epilepsy and valproic acid on oxidant status in children with idiopathic epilepsy. Epilepsy Res. 2009;84(2-3):232-7.
25. Arhan E, Serdaroglu A, Ozturk B, Ozturk HS, Ozcelik A, Kurt N, et al. Effects of epilepsy and antiepileptic drugs on nitric oxide, lipid peroxidation and xanthine oxidase system in children with idiopathic epilepsy. Seizure. 2011;20(2):138-42.
26. Kurekci AE, Alpay F, Tanindi S, Gokcay E, Ozcan O, Akin R, et al. Plasma trace element, plasma glutathione peroxidase, and superoxide dismutase levels in epileptic children receiving antiepileptic drug therapy. Epilepsia. 1995;36(6):600-4.
27. Yuksel A, Cengiz M, Seven M, Ulutin T. Changes in the antioxidant system in epileptic children receiving antiepileptic drugs: two-year prospective studies. J Child Neurol. 2001;16(8):603-6.
28. Ozden H, Kabay SC, Toker A, Ustuner MC, Ozbayer C, Ustuner D, et al. The effects of levetiracetam on urinary 15f-2t-isoprostane levels in epileptic patients. Seizure. 2010;19(8):514-6.
29. Bolayir E, Celik K, Tas A, Topaktas S, Bakir S. The effects of oxcarbazepine on oxidative stress in epileptic patients. Methods Find Exp Clin Pharmacol. 2004;26(5):345-8.
30. Martinc B, Grabnar I, Vovk T. Antioxidants as a preventive treatment for epileptic process: a review of the current status. Curr Neuropharmacol. 2014;12(6):527-50.
31. Higuchi S, Yano A, Takai S, Tsuneyama K, Fukami T, Nakajima M, et al. Metabolic activation and inflammation reactions involved in carbamazepine-induced liver injury. Toxicol Sci. 2012;130(1):4-16.
32. Cardenas-Rodriguez N, Huerta-Gertrudis B, Rivera-Espinosa L, Montesinos- Correa H, Bandala C, Carmona-Aparicio L, et al. Role of oxidative stress in refractory epilepsy: evidence in patients and experimental models. Int J Mol Sci. 2013;14(1):1455-76.
33. Brieger K, Schiavone S, Miller FJ, Krause KH. Reactive oxygen species: from health to disease. Swiss Med Wkly. 2012;142:w13659.
34. D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8(10):813-24.
35. Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7(1):65-74.
36. Mendez-Armenta M, Nava-Ruiz C, Juarez-Rebollar D, Rodriguez-Martinez E, Gomez PY. Oxidative stress associated with neuronal apoptosis in experimental models of epilepsy. Oxid Med Cell Longev. 2014;2014:293689.
37. Kann O, Kovacs R. Mitochondria and neuronal activity. Am J Physiol Cell Physiol. 2007;292(2):C641-57.
38. Ben-Menachem E, Kyllerman M, Marklund S. Superoxide dismutase and glutathione peroxidase function in progressive myoclonus epilepsies. Epilepsy Res. 2000;40(1):33-9.
39. Lopez J, Gonzalez ME, Lorigados L, Morales L, Riveron G, Bauza JY. Oxidative stress markers in surgically treated patients with refractory epilepsy. Clin Biochem. 2007;40(5-6):292-8.
40. Ho YH, Lin YT, Wu CW, Chao YM, Chang AY, Chan JY. Peripheral inflammation increases seizure susceptibility via the induction of neuroinflammation and oxidative stress in the hippocampus. J Biomed Sci. 2015;22:46.
41. Pecorelli A, Natrella F, Belmonte G, Miracco C, Cervellati F, Ciccoli L, et al. NADPH oxidase activation and 4-hydroxy-2-nonenal/aquaporin-4 adducts as possible new players in oxidative neuronal damage presents in drug-resistant epilepsy. Biochim Biophys Acta. 2015;1852(3):507-19.
42. Chuang YC, Chen SD, Lin TK, Liou CW, Chang WN, Chan SH, et al. Upregulation of nitric oxide synthase II contributes to apoptotic cell death in the hippocampal CA3 subfield via a cytochrome c/caspase-3 signaling cascade following induction of experimental temporal lobe status epilepticus in the rat. Neuropharmacology. 2007;52(5):1263-73.
43. Chuang YC, Chen SD, Liou CW, Lin TK, Chang WN, Chan SH, et al. Contribution of nitric oxide, superoxide anion, and peroxynitrite to activation of mitochondrial apoptotic signaling in hippocampal CA3 subfield following experimental temporal lobe status epilepticus. Epilepsia. 2009;50(4):731-46.
44. Henshall DC. Apoptosis signalling pathways in seizure-induced neuronal death and epilepsy. Biochem Soc Trans. 2007;35(Pt 2):421-3.
45. Vezzani A, Balosso S, Ravizza T. Inflammation and epilepsy. Handb Clin Neurol. 2012;107:163-75.
46. Lorigados PL, Morales CL, Orozco SS, Rocha AL. Pharmacoresistant epilepsy and immune system. In: Rocha AL, Cavalheiro EA, editors. Pharmacoresistance in Epilepsy. From Genes and Molecules to Promising Therapies. New York: Springer; 2013. p. 149-68.
47. Ureña-Guerrero ME, Feria-Velasco A, Gudiño-Cabrera G, Camin-Espuny A, Beas-Zárate C. Modifications in the Seizures Susceptibility by Excitotoxic Neuronal Damage and Possible Relationship with the Pharmacoresistance. In: Rocha AL, Cavalheiro EA, editors. Pharmacoresistance in Epìlepsy. From Genes and Molecules to Promising Therapy. NY: Springer; 2013. p. 59-76.
48. Lorigados L, Orozco S, Morales-Chacon L, Estupiñan B, García I, Rocha L. Excitotoxicity and neuronal death in epilepsy. Biotecnol Apl. 2013;30(1):9-16.
49. Vezzani A, Aronica E, Mazarati A, Pittman QJ. Epilepsy and brain inflammation. Exp Neurol. 2013;244:11-21.
50. Riazi K, Galic MA, Pittman QJ. Contributions of peripheral inflammation to seizure susceptibility: cytokines and brain excitability. Epilepsy Res. 2010;89(1):34-42.
51. Ravizza T, Balosso S, Vezzani A. Inflammation and prevention of epileptogenesis. Neurosci Lett. 2011;497(3):223-30.
52. Eid T, Tu N, Lee TS, Lai JC. Regulation of astrocyte glutamine synthetase in epilepsy. Neurochem Int. 2013;63(7):670-81.
53. Marchi N, Granata T, Janigro D. Inflammatory pathways of seizure disorders. Trends Neurosci. 2014;37(2):55-65.
54. Touyz RM, Deng LY, He G, Wu XH, Schiffrin EL. Angiotensin II stimulates DNA and protein synthesis in vascular smooth muscle cells from human arteries: role of extracellular signal-regulated kinases. J Hypertens. 1999;17(7):907-16.
55. De La Fuente M, Miquel J, Catalan MP, Victor VM, Guayerbas N. The amount of thiolic antioxidant ingestion needed to improve several immune functions is higher in aged than in adult mice. Free Radic Res. 2002;36(2):119-26.
56. Viora M, Quaranta MG, Straface E, Vari R, Masella R, Malorni W. Redox imbalance and immune functions: opposite effects of oxidized low-density lipoproteins and N-acetylcysteine. Immunology. 2001;104(4):431-8.
57. Uchida K, Shiraishi M, Naito Y, Torii Y, Nakamura Y, Osawa T. Activation of stress signaling pathways by the end product of lipid peroxidation. 4-hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. J Biol Chem. 1999;274(4):2234-42.
58. Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem. 2000;267(16):4912-6.
59. Wang X, Michaelis EK. Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci. 2010;2:12.
60. Streit WJ. Microglia and the response to brain injury. Ernst Schering Res Found Workshop. 2002(39):11-24.
61. Aronica E, Boer K, van Vliet EA, Redeker S, Baayen JC, Spliet WG, et al. Complement activation in experimental and human temporal lobe epilepsy. Neurobiol Dis. 2007;26(3):497-511.
62. Ravizza T, Gagliardi B, Noe F, Boer K, Aronica E, Vezzani A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis. 2008;29(1):142-60.
63. Boer K, Crino PB, Gorter JA, Nellist M, Jansen FE, Spliet WG, et al. Gene expression analysis of tuberous sclerosis complex cortical tubers reveals increased expression of adhesion and inflammatory factors. Brain Pathol. 2010;20(4):704-19.
64. Iyer A, Zurolo E, Spliet WG, van Rijen PC, Baayen JC, Gorter JA, et al. Evaluation of the innate and adaptive immunity in type I and type II focal cortical dysplasias. Epilepsia. 2010;51(9):1763-73.
65. Maldonado M, Baybis M, Newman D, Kolson DL, Chen W, McKhann G, 2nd, et al. Expression of ICAM-1, TNF-alpha, NF kappa B, and MAP kinase in tubers of the tuberous sclerosis complex. Neurobiol Dis. 2003;14(2):279-90.
66. Bauer S, Cepok S, Todorova-Rudolph A, Nowak M, Koller M, Lorenz R, et al. Etiology and site of temporal lobe epilepsy influence postictal cytokine release. Epilepsy Res. 2009;86(1):82-8.
67. Eid T, Williamson A, Lee TS, Petroff OA, de Lanerolle NC. Glutamate and astrocytes- -key players in human mesial temporal lobe epilepsy? Epilepsia. 2008;49 Suppl 2:42-52.
68. Arundine M, Tymianski M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium. 2003;34(4-5):325-37.
69. Dong XX, Wang Y, Qin ZH. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin. 2009;30(4):379-87.
70. Sloviter RS. The neurobiology of temporal lobe epilepsy: too much information, not enough knowledge. C R Biol. 2005;328(2):143-53.
71. Liang LP, Ho YS, Patel M. Mitochondrial superoxide production in kainate-induced hippocampal damage. Neuroscience. 2000;101(3):563-70.
72. Shin EJ, Jeong JH, Chung YH, Kim WK, Ko KH, Bach JH, et al. Role of oxidative stress in epileptic seizures. Neurochem Int. 2011;59(2):122-37.
73. Jarrett SG, Liang LP, Hellier JL, Staley KJ, Patel M. Mitochondrial DNA damage and impaired base excision repair during epileptogenesis. Neurobiol Dis. 2008;30(1): 130-8.
74. Waldbaum S, Patel M. Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res. 2010;88(1):23-45.
75. Waldbaum S, Liang LP, Patel M. Persistent impairment of mitochondrial and tissue redox status during lithium-pilocarpine-induced epileptogenesis. J Neurochem. 2010;115(5):1172-82.
76. Dal-Pizzol F, Klamt F, Vianna MM, Schroder N, Quevedo J, Benfato MS, et al. Lipid peroxidation in hippocampus early and late after status epilepticus induced by pilocarpine or kainic acid in Wistar rats. Neurosci Lett. 2000;291(3):179-82.
77. Cini M, Moretti A. Studies on lipid peroxidation and protein oxidation in the aging brain. Neurobiol Aging. 1995;16(1):53-7.
78. Bruce AJ, Baudry M. Oxygen free radicals in rat limbic structures after kainate-induced seizures. Free Radic Biol Med. 1995;18(6):993- 1002.
79. Frantseva MV, Perez Velazquez JL, Tsoraklidis G, Mendonca AJ, Adamchik Y, Mills LR, et al. Oxidative stress is involved in seizure-induced neurodegeneration in the kindling model of epilepsy. Neuroscience. 2000;97(3):431-5.
80. Tejada S, Sureda A, Roca C, Gamundi A, Esteban S. Antioxidant response and oxidative damage in brain cortex after high dose of pilocarpine. Brain Res Bull. 2007;71(4):372-5.
81. Folbergrova J, Kunz WS. Mitochondrial dysfunction in epilepsy. Mitochondrion. 2012;12(1):35-40.
82. Bellissimo MI, Amado D, Abdalla DS, Ferreira EC, Cavalheiro EA, Naffah-Mazzacoratti MG. Superoxide dismutase, glutathione peroxidase activities and the hydroperoxide concentration are modified in the hippocampus of epileptic rats. Epilepsy Res. 2001;46(2):121-8.
83. Patel M, Liang LP, Roberts LJ, 2nd. Enhanced hippocampal F2-isoprostane formation following kainate-induced seizures. J Neurochem. 2001;79(5):1065-9.
84. Kudin AP, Kudina TA, Seyfried J, Vielhaber S, Beck H, Elger CE, et al. Seizure-dependent modulation of mitochondrial oxidative phosphorylation in rat hippocampus. Eur J Neurosci. 2002;15(7):1105-14.
85. Waldbaum S, Patel M. Mitochondrial dysfunction and oxidative stress: a contributing link to acquired epilepsy? J Bioenerg Biomembr. 2010;42(6):449-55.
86. Sudha K, Rao AV, Rao A. Oxidative stress and antioxidants in epilepsy. Clin Chim Acta. 2001;303(1-2):19-24.
87. Vielhaber S, Niessen HG, Debska-Vielhaber G, Kudin AP, Wellmer J, Kaufmann J, et al. Subfield-specific loss of hippocampal N-acetyl aspartate in temporal lobe epilepsy. Epilepsia. 2008;49(1):40-50.
88. Heller A, Kunz M, Samakas A, Haase M, Kirschfink M, Koch T. The complement regulators C1 inhibitor and soluble complement receptor 1 attenuate acute lung injury in rabbits. Shock. 2000;13(4):285-90.
89. Turkdogan D, Toplan S, Karakoc Y. Lipid peroxidation and antioxidative enzyme activities in childhood epilepsy. J Child Neurol. 2002;17(9):673-6.
90. Aycicek A, Iscan A. The effects of carbamazepine, valproic acid and phenobarbital on the oxidative and antioxidative balance in epileptic children. Eur Neurol. 2007;57(2):65-9.
91. Verrotti A, Basciani F, Trotta D, Pomilio MP, Morgese G, Chiarelli F. Serum copper, zinc, selenium, glutathione peroxidase and superoxide dismutase levels in epileptic children before and after 1 year of sodium valproate and carbamazepine therapy. Epilepsy Res. 2002;48(1-2):71-5.
92. Gunes S, Dirik E, Yis U, Seckin E, Kuralay F, Kose S, et al. Oxidant status in children after febrile seizures. Pediatr Neurol. 2009;40(1):47-9.
93. Lorigados Pedre L, Orozco Suarez S, Morales Chacon L, Garcia Maeso I, Estupinan Diaz B, Bender del Busto JE, et al. Neuronal death in the neocortex of drug resistant temporal lobe epilepsy patients. Neurologia. 2008;23(9):555-65.
94. Lorigados Pedre L, Morales Chacon LM, Orozco Suarez S, Pavon Fuentes N, Estupinan Diaz B, Serrano Sanchez T, et al. Inflammatory mediators in epilepsy. Curr Pharm Des. 2013;19(38):6766-72.
95. Estupinan-Diaz B, Morales-Chacon LM, Lorigados-Pedre L, Garcia-Maeso I, Bender-del Busto JE, Trapaga-Quincoses O, et al. Pathological neocortical findings in patients with medication-resistant medial temporal lobe epilepsy submitted to surgery. Rev Neurol. 2008;46(4):203-9.
96. Vatassery GT. Vitamin E. Neurochemistry and implications for neurodegeneration in Parkinson’s disease. Ann N Y Acad Sci. 1992;669:97-109.
97. Pisoschi AM, Pop A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur J Med Chem. 2015;97:55-74.
98. Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012;5(1):9-19.
99. Naziroglu M, Yurekli VA. Effects of antiepileptic drugs on antioxidant and oxidant molecular pathways: focus on trace elements. Cell Mol Neurobiol. 2013;33(5):589-99.
100. Freitas RM. The evaluation of effects of lipoic acid on the lipid peroxidation, nitrite formation and antioxidant enzymes in the hippocampus of rats after pilocarpine-induced seizures. Neurosci Lett. 2009;455(2):140-4.
101. Cardenas-Rodriguez N, Coballase-Urrutia E, Huerta-Gertrudis B, Garcia-Cruz ME, Pedraza-Chaverri J, Coria-Jimenez R, et al. Antioxidant activity of topiramate: an antiepileptic agent. Neurol Sci. 2013;34(5):741-7.
102. Niedzielska E, Smaga I, Gawlik M, Moniczewski A, Stankowicz P, Pera J, et al. Oxidative Stress in Neurodegenerative Diseases. Mol Neurobiol. 2016;53(6):4094-125.
103. Rong Y, Doctrow SR, Tocco G, Baudry M. EUK-134, a synthetic superoxide dismutase and catalase mimetic, prevents oxidative stress and attenuates kainate-induced neuropathology. Proc Natl Acad Sci U S A. 1999;96(17):9897-902.
104. Gupta YK, Gupta M, Kohli K. Neuroprotective role of melatonin in oxidative stress vulnerable brain. Indian J Physiol Pharmacol. 2003;47(4):373-86.
105. Kong Q, Lin CL. Oxidative damage to RNA: mechanisms, consequences, and diseases. Cell Mol Life Sci. 2010;67(11):1817- 29.
106. Ambrogini P, Minelli A, Galati C, Betti M, Lattanzi D, Ciffolilli S, et al. Post-seizure alpha-tocopherol treatment decreases neuroinflammation and neuronal degeneration induced by status epilepticus in rat hippocampus. Mol Neurobiol. 2014;50(1):246-56.
107. Holtzman D, Khait I, Mulkern R, Allred E, Rand T, Jensen F, et al. In vivo development of brain phosphocreatine in normal and creatine-treated rabbit pups. J Neurochem. 1999;73(6):2477-84.
108. Mori A, Noda Y, Packer L. The anticonvulsant zonisamide scavenges free radicals. Epilepsy Res. 1998;30(2):153-8.
109. Lima PA, Sampaio LP, Damasceno NR. Neurobiochemical mechanisms of a ketogenic diet in refractory epilepsy. Clinics (Sao Paulo). 2014;69(10):699-705.
110. Auvin S. Fatty acid oxidation and epilepsy. Epilepsy Res. 2012;100(3):224-8.
111. Giordano C, Marchio M, Timofeeva E, Biagini G. Neuroactive peptides as putative mediators of antiepileptic ketogenic diets. Front Neurol. 2014;5:63.
112. Jarrett SG, Milder JB, Liang LP, Patel M. The ketogenic diet increases mitochondrial glutathione levels. J Neurochem. 2008;106(3):1044-51.
113. Milder J, Patel M. Modulation of oxidative stress and mitochondrial function by the ketogenic diet. Epilepsy Res. 2012;100(3):295-303.
114. Zamani GR, Mohammadi M, Ashrafi MR, Karimi P, Mahmoudi M, Badv RS, et al. The effects of classic ketogenic diet on serum lipid profile in children with refractory seizures. Acta Neurol Belg. 2016;116(4):529-34.
Received in February, 2016.
Accepted in June, 2016.
Lourdes Lorigados. Departamento de Inmunoquímica, Centro Internacional de Restauración Neurológica, Ciren. Ave. 25 No. 15805 e/ 158 y 160, CP 11300, Playa, La Habana, Cuba. E-mail: lourdesl@neuro.ciren.cu.
REVIEW


{kind=link}
{kind=link}