










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBiotecnología Aplicada
versión On-line ISSN 1027-2852
Biotecnol Apl vol.33 no.2 La Habana abr.-jun. 2016
RESEARCH
Real-time stability of the Ecuadorian pentavalent DTP-HB-Hib vaccine
Estudio de estabilidad real de la vacuna pentavalente DPT-HB-Hib ecuatoriana
Néstor Expósito1, Gladys Alvarez2, Virginia Riera2, Jenny Navas2, Mabel Izquierdo1, Adela Andrade2, Blanca Nuñez1, Diana Sanchez2, Beatriz Brito2, Zoe Ñuñez1, Annette Pereira1, Yamila Martínez1, Yayrí Prieto1
1 Dirección de Desarrollo Tecnológico, Centro de Ingeniería Genética y Biotecnología, CIGB. Ave. 31 e/ 158 y 190, Cubanacán, Playa, CP 11600, La Habana, Cuba.
2 Producción de Biológicos, Empresa Pública de Fármacos, ENFARMA EPAv. Julián Coronel 905 entre Esmeraldas y José Mascote, Guayaquil, Ecuador.
ABSTRACT
The Ecuadorian pentavalent vaccine consists of five antigens: diphtheria and tetanus toxoid, whole-cell Bordetella pertussis, the hepatitis B surface antigen, and the Hemophilus influenzae polyribosyl ribitol phosphate conjugated to the tetanus toxoid (DPT-HB-Hib). Previously, a preformulation study was carried out, with the technology required for the formulation of the vaccine defined. Therefore, in this work, a stability study was conducted over 36 months. Seventy two samples from five batches in 6R multidose vials packed in cardboard boxes were stored at 5 ± 3 °C. They were evaluated at seven time points (0, 6, 12, 18, 24, 30 and 36 months) for organoleptic properties, thimerosal and aluminum content, pH, toxicological assays, sterility and antigenic identity and potency, as recommended for combination vaccines. The results of this long-term evaluation allowed us to define a shelf-life of 30 months for the vaccine, supported by the potency and immunogenicity of the five antigens and the toxicity of the vaccine which met the specifications established for this product. This result gives us the coverage for batches manufactured for clinical tests to evaluate safety and efficacy in humans.
Keywords: Pentavalent vaccine, real-time stability, vaccine potency, vaccine immunogenicity
RESUMEN
La vacuna pentavalente ecuatoriana está constituida por cinco antígenos: anatoxinas diftérica y tetánica, células enteras de Bordetella pertussis, el antígeno de superficie del virus de la hepatitis B recombinante y el polirribosilribitol fostato conjugado a la anatoxina tetánica (DPT-HB-Hib). Para tales propósitos, las 72 muestras de cinco lotes envasados en viales multidosis 6R y estuches de cartón fueron almacenadas a 5 ± 3 °C. Estas fueron evaluadas en siete puntos temporales (0, 6, 12, 18, 24, 30 y 36 meses), para los parámetros de pH, ensayos toxicológicos, de esterilidad y de identidad y potencia antigénicas, acordes con las recomendaciones para las vacunas combinadas. Los resultados obtenidos a largo plazo durante la evaluación de la estabilidad por 36 meses permitió definir un tiempo de vida para la vacuna de 30 meses, basado en las determinaciones de la potencia y la inmunogenicidad de los cinco antígenos y la toxicidad de la vacuna, parámetros todos que satisfacen las especificaciones establecidas para este tipo de producto. Este resultado nos facilita una cobertura de utilización de los lotes que se fabriquen para los estudios clínicos, con vistas a evaluar su seguridad y eficacia en seres humanos.
Palabras clave: vacuna pentavalente, estabilidad en tiempo real, potencia vacunal, inmunogenicidad vacunal.
INTRODUCTION
The creation of the Extended Immunization Program has enabled the vaccination of hundreds of thousands of persons in the world with the application of millions of dosages of vaccines, in places where there are inappropriate storage conditions. For this, the World Health Organization (WHO) defined the need for specific studies on vaccine stability and to collect all the existent information [1-3].
Stability is defined as the ability of a pharmaceutical product to preserve its chemical, physical, microbiological and biopharmaceutical properties within specified limits, throughout its preservation period [4].
Vaccines are a heterogeneous class of preparations containing proteins, polysaccharides, anatoxins, DNA recombinant antigens, inactivated viruses or live attenuated microorganisms as their active components. They can also contain stabilizers, adjuvants, preservatives and other excipients, which all contribute to their quality, safety and efficacy [5].
The results of vaccine or drug stability studies help establish their expiry dates, indicating the maximum validity time, thus ensuring the potency, purity, physicochemical characteristics and others that correspond to the nature and indication of the drug [6].
Historically, the vaccine stability studies were based on the sensitivity observed in the results at different temperatures. This led to the design of the need of a cold chain and storage. During the 1980’s, thermostability studies were introduced as a requirement for the use of vaccines such as rabies and measles-mumps-rubella [7].
More recently, WHO has pointed out the importance of stability studies for vaccines stored in real time [8]. It is currently known that a vaccine can lose efficacy when exposed to temperatures below 0 ºC, as in the case of vaccines adsorbed onto aluminum adjuvants, since the adjuvant losses its immune antigen response potentiating property during freezing [9]. Moreover, accidental interruptions of the cold chain affect vaccine stability, efficacy and safety at variable degrees depending on their characteristics, the maximum or minimum temperatures recorded and the exposure time to that temperature. This could affect their pharmacological properties and produce important economic losses for the health system, given the high cost of some of these drugs [10].
During the specific development of combined vaccines, besides achieving a technologically consistent, stable and appropriate formulation, the big problem is quality control; it’s a tool that must show that the process is consistent, and that the product has quality, stability, efficacy and safety [11-13]. This is very costly because of the diverse tests required, which may in some cases be different from the ones used for each separate component, since some antigens interact with others, with the preservative, with the adjuvant and other additives, altering the original indicators [14, 15].
The tests carried out in vaccine stability studies include those of potency and immunogenicity; however, this may be a problem since they must show that the combined antigens have biological activity that is at least equal to that reached when administered separately [16, 17]. On the other hand, biological tests have low precision and high variability because of the use of animals, as well as the complexity of tests such as the challenges that are made throughout the stability study. These studies must follow specific quality specifications, which include the references of the analytical procedures, the appropriate acceptance criteria, according to the specific elements, with numerical, qualitative or other types of limits. A series of other additionally relevant information, depending on the type of element or product, is also needed; this may cover components, intermediate products, finished products or other products or materials required [18].
Therefore, this work was aimed to evaluate the stability study of a pentavalent vaccine produced in Ecuador [19], composed by the diphtherial and tetanus anatoxins (D and T, respectively), and whole cells of Bordetella pertussis (P) produced in Ecuador, and the recombinant hepatitis B virus surface antigen (HBsAg) and the synthetic polysaccharide (polyribosyl ribitol phosphate, PRP) of Haemophilus influenzae conjugated to tetanus anatoxin (PRP-T) produced at the CIGB. The showed positive results obtained in the physicochemical and biological tests in real-time stability testing enabled us to establish the validity period of this Ecuadorian pentavalent vaccine to 30 months.
MATERIALS AND METHODS
Formulation of vaccine batches
For the real-time stability studies of the Ecuadorian pentavalent vaccine, three batches at 10-L scale each were formulated and filled. They were formulated by diluting the phosphate aluminum gel from 2 to 1 % with saline solution at 0.85 %, pH 6.6. The total volume of the gel was divided into two flasks (flask 1 and flask 2); both were sterilized at 121 °C for 15 min.
The sterile purified diphtherial anatoxin was poured into flask 1 at room temperature, by using a magnetic shaker (IKA, RCT Basic, Germany), at 200 rpm for 2h30min. Afterwards, the tetanus anatoxin was added; it was shaken for 2h30min. Then, the whole cells of B. pertussis were added, while slowly shaking for 18 h.
The PRP-T was placed in flask 2; the adsorption process took place while shaking with the magnetic shaker (IKA, RCT Basic, Germany) at 200 rpm; shaking was slow for 5 h and later the HBsAg was added and the adsorption process took place during 18 h.
Both flasks were then mixed and the buffer solution of 8 mM PBS and thimerosal were added.
The batches were dispensed at 5 mL per vial where each dose is of 0.5 mL. The batches were named VPE 1003, VPE 1004 and VPE 1005.
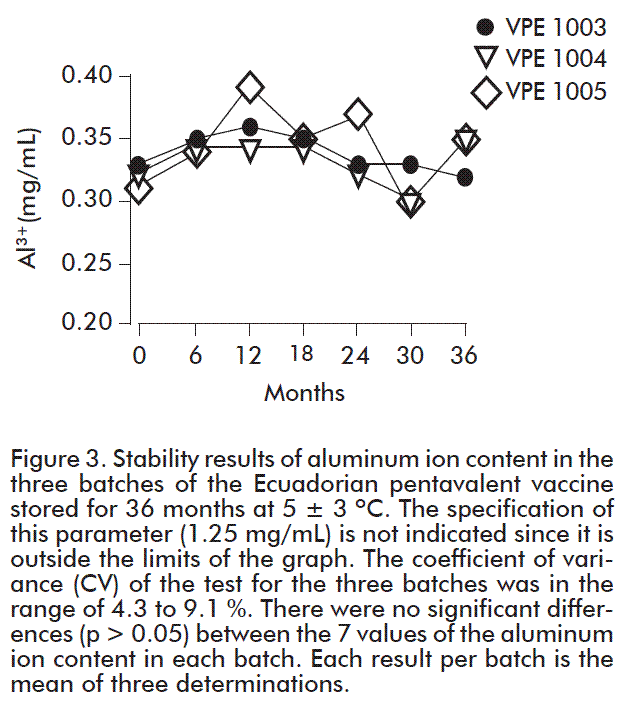
The conformation of the batches of the pentavalent vaccine was made using different batches of the API of D, P, T, HBsAg and PRP-T as shown in Table 1.
Vial sealing system
6R hydrolytic No. 1 boron silicate vials were filled, and Butyl rubber stoppers and aluminum seals of 20 mm were used to plug them. A self-adhesive label was placed on each vial with the information regarding the vaccine, and the secondary packaging consisted of white cardboard cases.
Sampling frequency
According to the ICH Q1A guidelines (R2), the refrigerated products must comply with a true stability testing program covering the entire study at a storage temperature of 5 ± 3 °C for 12 months and an accelerated study where the samples are stored at 25 ± 2 °C and 60 ± 5 % of relative humidity (RH) for 6 months [20].
A design was established where the storage temperature of the vials of the three lots of the Ecuadorian pentavalent vaccine was of 5 ± 3 °C for 36 months, to guarantee a true stability study in compliance with guidelines. The cold chamber temperature where the batches were stored was monitored 3 times a day throughout the study period.
Recommendations establish a sampling frequency every 3 months in the first year of storage of the vaccine, every 6 months during the second year of storage, and once a year in the third year of storage [20]. The sampling frequency was defined in the following way: all physicochemical and biological tests, as well as those of potency and immunogenicity of the vaccine were made at time 0 (the release of the batch) and at 6, 12, 18, 24, 30 and 36 months. The potency tests of the antigen lasted for up to three months; 7 sampling time points were set since the results at months 3 and 9 months overlapped.
A matrix sampling plan was used to carry out the test since a fraction of the total number of vials per batch was tested, corresponding to the 7 sampling points mentioned above. The number of vials per test were as follows: sterility, 20 vials; general toxicity, 4; diphtherial anatoxin potency, 5; tetanus anatoxin potency, 5; B. pertussis potency, 6; HBsAg potency, 3; immunogenicity of the PRP-T, 2; specific toxicity of the anatoxins, 10; toxicity of B. pertussis, 5; identity of HBsAg, 2; identity of PRP-T, 2; pH, 2; thimerosal, 2; and the aluminum ion content, 6. Overall, at each sampling point (0, 6, 12, 18, 24, 30 and 36 months) a total of 72 vials were taken from each batch for the corresponding tests.
In the case of the organoleptic features of the samples, the tests were not destructive and samples were reincorporated into the batch. The design assumed that the stability of the samples tested represented the stability of all samples of the lot.
Physicochemical tests
Determination of organoleptic characteristics
Each lot was sampled to evaluate organoleptic characteristics; a batch was considered to pass the test if the vaccine was observed as a grayish liquid that was free from particles, which separated into two phases when at rest, with a gray white sediment (adjuvant) and a transparent supernatant that were readily resuspended when shaking [21, 22].
pH
This method was based on the potentiometric determination of the hydrogen ion concentration in the product, measured through the use of electrodes and a Mettler Toledo Seven Easy pHmeter. The range of the proposed specification for this test was 6.4 to 7.4 [23, 24].
Thimerosal content
Thimerosal was indirectly determined in the vaccine tested by mercury spectrophotometric quantification, which forms a complex with the dithizone reagent. Separation was carried out through its extraction with chloroform. The specification proposed for this parameter is of 0.005-0.02 g% of thimerosal per milliliter of the vaccine [25].
Aluminum ion content
The test used was based on the indirect determination of the aluminum phosphate through the quantification of the Al3+ located in its molecule. The Al3+ forms a complex with the EDTA in an acetate regulating solution, valuating the excess of EDTA with pentahydrated copper sulfate II in the presence of the indicating solution of 1-pyridyl-2 azonaphthol. The vaccine was regarded as passing the test if the Al3+ ion content was equal or less than 1.25 mg per single human dose (SHD) [26].
Toxicological tests
General safety
This test was carried out according to requirements of the British Pharmacopoeia, and its aim was to determine toxic reactions and weight loss in mice and guinea pigs after the intraperitoneal injection of 0.5 mL of the test sample per animal. The batch passes the test when no animal dies or shows symptoms of disease [21, 27].
Specific toxicity of anatoxins and B. pertussis
Diphtherial anatoxin
The test is based on the observation of the signs shown by the animals inoculated with the tested samples, when free toxin is present because of a deficient detoxification or an anatoxin reversion process.
There were five guinea pigs of the same sex weighing 250-350 g in the test. Each one was inoculated by the intramuscular route with 5 mL of the vaccine, divided into two applications of 2.5 mL each. The animals were observed daily in the search for signs of intoxication with diphtherial toxin. The control group used purified diphtherial anatoxin.
The vaccine lot passes the test if no animal shows the characteristic signs of diphtheria for 6 weeks after the inoculation date, and if at least 80 % of the animals survive the test period [28].
Tetanus anatoxin
The same procedure was described above was followed, where the control group used purified tetanus anatoxin. The batches passed the test if no animal showed symptoms of specific paralysis or any other sign of tetanus within 4 weeks after the inoculation and if at least 80 % of the animals survived the test period [28].
Specific toxicity of B. pertussis
To evaluate the batches, 10 OF-1 mice of the same sex weighing 14-16 g were used for each sample, and a physiological saline solution was used for the control. The mice were injected by the intraperitoneal route with 0.5 mL of the vaccine being tested and were later observed [29].
The batch passed the test if:
- After 72 h the total weight of the group should not be less than the weight before the injection.
- At the end of the seventh day, the average weight gain per mouse cannot be less than 60 % of that of the control group of mice.
- At the end of the test the death of the injected mice cannot be more than 5 %.
Sterility
The sterility test was made according to the requirements of the USP 30 and it was based on microbial growth in the thioglycolate and tryptone-soy agar culture media. If no contamination was observed after 14 days, it was determined that the sample passed the test in a satisfactory manner [23, 30].
Identity tests
Identity of the diphtherial and tetanus anatoxins
For this test we used the Ramon’s identification method, by mixing variable amounts of the anatoxin with constant amounts of the antitoxin under permanent observation and constant temperature. The mixture flocculating the first indicates the approximate amount of the anatoxin found in the sample, which is expressed as the limit of flocculation (Lf). The test quantified the amount of anatoxin that produces a white precipitate in the shortest time possible, when mixed with 1 IU of the antitoxin [31, 32].
Identity of B. pertussis
The presence of the cells of B. pertussis is evaluated in formulations using specific sera against the 3 agglutinogens, pertussis toxin, filamentous hemagglutinin and pertactin, obtained in the laboratory after immunizing the rabbits with the purified antigens.
The pentavalent vaccine samples were mixed with sodium citrate at a concentration of 100 g/L, and later stored at 37 °C for 16 h; after that time they were centrifuged at 2000 rpm for 15 min to obtain a transparent supernatant liquid. From this supernatant, 50 μL were taken and mixed them in U-bottom plates (Nunc Maxisorp) at serial dilutions with a factor of 2 of the specific sera previously diluted 1:4.
The plates were incubated for 24 h at 37 °C in a humid chamber. A positive agglutination well was that in which an agglutination clumping was formed. A negative control was incorporated that did not include the serum [33].
Identity of the HBsAg
The identity test of the HBsAg was made in two stages: the separation of the HBsAg of the adjuvant gel and the identification of the antigen by ELISA.
Five milliliters of the tested vaccine were taken and 400 mM phosphate buffer saline (PBS) were added and shaken in a vortex (IKA MS1, Germany) for 1 min to deabsorpt the HBsAg from the adjuvant. Afterwards, 1 mL of the sample was centrifuged (Sigma 204, Germany) at 3500 rpm for 5 min; then, 200 μL of the supernatant was collected and the ELISA was carried out by coating the plates with 100 μL of a goat polyclonal antibody against HBsAg (006). The plates were incubated for 15 min at 50 ºC, and further washed three times with PBS- 0.05 % Tween 20. After washing, 200 μL were taken from the standard solution and from the vaccine, and diluted 2 times with 7 mM PBS.
Subsequently, the samples were applied on the plate, incubated for 4 hours at 37 ºC in a humid chamber, washed as previously mentioned and 100 μL of the anti-sheep peroxidase conjugate (1/100) were added to the plate. They were incubated for 1 h at 50 ºC, washed with 100 μL of the buffer solution of citrate-phosphate, containing the substrate of 1 mg/mL ortho-phenylendiamine and 30 % H2O2. The reaction was stopped by adding 50 μL of the stopping solution (2 M H2SO4); the plate was read at 492 nm in a Multiskan Plus “Titertek”. The result was positive if the HBsAg was detected in the vaccine tested [34].
Identity of the PRP-T
The qualitative method of immuno-identification by latex was used through a set of commercial reagents from Pastorex® Meningitis (61716). For the determination, a drop of the well homogenized vaccine was taken with a Pasteur pipette and mixed with a drop of latex sensitized with rabbit monoclonal antibodies that were specifically against the capsular polysaccharide of the H. influenzae type b bacterium. The test was considered positive if the sample agglutinated the positive control in the same way [35].
Biological tests
Potency of the diphtherial and tetanus anatoxins according to the indirect method (FDA)
This procedure is based on the neutralization capacity of diphtherial and tetanus anatoxins found in the sera mixture of animals that had been immunized with the pentavalent vaccine, compared to the diphtherial and tetanus reference toxins.
Through the subcutaneous route, 6 Swiss albino guinea pigs of 450-500 g weight were immunized with 0.25 mL of the pentavalent vaccine (half of the total human immunizing dose). The animals were bled four to six weeks later and the test tubes were incubated at 37 °C for 2 h, after which the clots were separated from the walls of the test tubes and refrigerated at 5 ± 3 °C for clot retraction.
An equal amount of serum was taken from each animal and placed in one test tube (sera pool). For diphtherial anatoxin titration, the diphtherial anatoxin reference was diluted up to 1.0 IU/mL with a physiologic saline solution.
From the mixture of animal sera, 0.75 mL and 1.5 mL were taken and 5.25 mL and 4.5 mL of the gelatin buffer solution were diluted to 2 and 4 arbitrary units per milliliter (AU/mL), respectively.
A total of 0.5 mL of the diphtherial toxin was taken and diluted with 4.5 mL of the gelatin buffer solution; this operation was repeated in a test tube containing 9 mL of the gelatin buffer solution.
The mixtures were prepared with 3 mL of the diphtherial toxin and with the dilution of the serum-gelatin buffer solution; they were then maintained in the dark at room temperature for 1 h.
Two albino guinea pigs of 250-300 g were inoculated with 3 mL of each preparation through the subcutaneous route. They were observed daily for a week to evaluate symptoms and the death of animals due to diphtheria, and the IU of diphtherial antitoxin per milliliter of serum were detected.
The lot passed the test when reaching 2 or more IU of diphtherial antitoxin per milliliter of serum [36].
Tetanus anatoxin titration was carried out as follows. The standard tetanus antitoxin was diluted to 0.1 IU/mL with a physiologic saline solution. From the immunized guinea pig sera pool, 1 mL was taken and diluted with 9 mL of the physiologic saline solution. Then, 1.5 mL and 0.75 mL of the dilution of sera of guinea pigs were taken and diluted with 4.5 and 5.25 mL of the gelatin-buffer solution to 4 and 2 AU/mL, respectively.
Subsequently, 0.5 mL of the tetanus toxin were taken and diluted with 4.5 mL of the gelatin-buffer solution; this operation was repeated in a test tube containing 9 mL of the gelatin-buffer solution.
The mixtures were prepared with 3 mL of tetanus toxin and with the serum-gelatin buffer solution; they were kept in the dark at room temperature for 1 h. Two albino guinea pigs of 350-400 g were inoculated with 3 mL of each preparation by the subcutaneous route.
They were observed daily for a week. The symptoms and death of the animals due to tetanus were observed and the IU of the tetanus antitoxin per milliliter of the serum was determined.
The batch passed the test if it reached 2 IU or more of the tetanus antitoxin per milliliter of the serum [36].
Potency of B. pertussis using the WHO method
The pertussis potency of the vaccine was determined by the comparison of a working reference vaccine approved by the Quality Control Division of the Finlay Institute (Havana, Cuba; Reference lot VPR(1)/99), which was calibrated against the international standard for the pertussis vaccine.
Four dilutions of the reference vaccine and of each vaccine batch to be tested were made. The serial dilutions were prepared with a dilution factor that was no larger than five, for which a sterile 0.85 % sodium chloride saline solution was used.
Albino OF-1 mice of 10-18 g of weight were injected intraperitoneally with 0.5 mL of the dilution corresponding to each mouse in each immunization group. Afterwards, the mice that had been immunized with the reference vaccine and the test vaccine were injected with the challenge dose by the intra-cerebral route at a time interval of 14-17 days after the immunization. The strain used for the challenge was B. pertussis 18 323.
To obtain estimates of the half lethal dose (LD50), dilutions of the challenge dose were made (1:50, 1:250, 1:1 250) that were inoculated within the brain of groups of control mice. The appropriate dilutions of the challenge dose were grown in a Bordet-Gengou agar base to determine the number of colony forming units (c.f.u.). The mice were observed for 14 days, and those dying within the first 72 h after the inoculation were excluded from the test.
The mice dying after 72 h of the inoculation were recorded to determine the half effective dose (ED50) of the vaccines. ED50 for each preparation was determined by the Probit statistical method [37] that assesses the linearity of the dose-response and the parallelism of the behavior of the vaccine in the test with the reference vaccine. The value of the ED50 of each vaccine was set as the intermediate value between the highest and lowest immunizing dose and the regressions that did not show significant linearity and parallelism (p ≤ 0.05). The challenge dose contained 100 to 1000 LD50 and no more than 300 c.f.u. The ED50 of the vaccine in the test and the standard vaccine were calculated by a method that offers an estimate of the limits of the 95 % confidence interval. The potency was estimated in terms of IU in the volume recom-mended for SHD.
The test vaccine complied with requirements for potency if the result of the test was statistically valid, showing that the estimated potency of the vaccine was not less than 4.0 IU per SHD [38].
Determination of the in vivo relative potency of the HBsAg
The potency test of the HBsAg was carried out according to the technical requirements of WHO [39]. For this, 1 mL of the test vaccine containing 20 μg/mL of HBsAg was diluted 1:16, 1:64, 1:256, 1:512 and 1:1 024 with aluminum phosphate gel at a concentration of 0.5 mg of Al+3/mL.
Ten female mice of 5 to 6 weeks of age and of the Balb/c, haplotype H-2d,q per group were immunized by the intraperitoneal route. Three batches of the pentavalent vaccine, one batch of the placebo and the reference batch of the vaccine against hepatitis B, 07-0902, were studied.
Twenty-eight days later, the mice were bled by retro-orbital puncture and the response of anti-HBsAg antibodies was assessed by ELISA, coating the plates with HBsAg (solid phase). The sample was incubated in the wells of the plate and then HBsAg conjugated with horseradish peroxidase was added. Ortho-phenylendiamine was used as the chromogenic substrate to develop the reaction. The ELISA plate was read at 492 nm.
The batch passed the test if the value of the relative potency was equal or higher than 0.5 compared to the potency of the reference vaccine [40].
Determination of immunogenicity of the PRP-T component
Groups of five F1 rabbits were immunized by the subcutaneous route with doses of 0.5 mL of the pentavalent vaccine corresponding to 10 μg of PRP-T. At the same time, rabbits were immunized with a control vaccine against Hib (Vaxem Hib, Chiron S.p.a, batch 3204) and the negative control was an aluminum phosphate placebo; 0.5 mg/mL.
The rabbits were immunized on days 0 and 14 and they were bled at 21 days after the first dose. Blood was collected individually.
The antibody response against PRP-T was assessed through a specific non-competitive and indirect ELISA system. Coating was performed according to international recommendations: a capsular polysaccharide of the bacteria was conjugated covalently to human serum albumin (HbO-HA; NIBSC, England). The sample was included to form the HbO-HA+Ac anti-PRP-T complex. This complex was bound to the conjugated mouse anti-IgG marked with peroxi-dase. Orthophenylendiamine was used as the chromogen and the reaction substrate was H2O2. A yellow-orange color appeared in the antibody positive samples.
The percentage of animals showing seroconversion for each dose was calculated for each test sample; the batch passed the test when seroconversion was found in at least 50 % of the animals immunized per study group and the average titer for each group of rabbits immunized with the pentavalent vaccine had to be equal or higher than 800 IU/mL [41].
Statistical analysis
The statistical significance of the values of the tests was analyzed by simple regression tests by analysis of variance (ANOVA) for a confidence level of 95 %. The differences between the results were considered to be significant when the p value was less than 0.05 (p < 0.05). The coefficient of variation (CV) of the tests was used for the comparison with data from the literature. The statistical program Statgraphics Centurion XV.II was used for these analyses.
RESULTS AND DISCUSSION
Physicochemical characterization of the stability of the Ecuadorian pentavalent vaccine
Organoleptic characteristics
The inspection of the three batches made during the seven sampling points showed that the vaccine was a suspension. When at rest, it separated into two phases, a transparent supernatant and a precipitated fraction corresponding to the adjuvant gel that, when gently shaken, it readily resuspended. The color of the vaccine remained constant as of the release of the batches up to 36 months after the end of the study and no lumps were formed and no foreign particles observed, for which we considered the result as satisfactory.
The material used for bottling and for secondary packing will also be used for marketing the Ecuadorian pentavalent vaccine after once registered.
pH
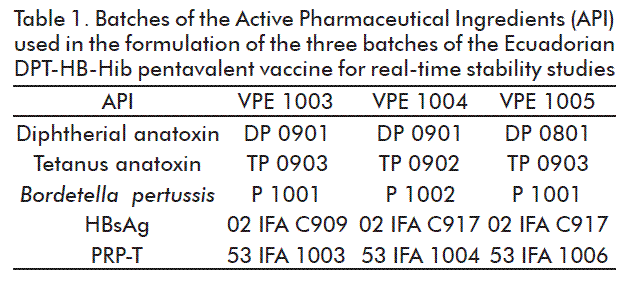
The results of the stability of the pH of the Ecuadorian pentavalent vaccine are shown in Figure 1. The pH values throughout the study were in the range 6.5-6.8, showing 0.7 to 1.5 % variability up to 36 months in the 3 batches studied. The values reported during the study for each batch showed no significant statistical differences (p > 0.05) with a 95 % confidence level. The pH defined for this vaccine was in the range of 6.4-7.4. The formulation process the pH of the vaccine was adjusted to the lowest specification limit, since the vaccine is intended to be administered through the intramuscular route and the pH of the intracellular and extracellular is 7.0 and 7.4, respectively. Moreover, based in our experience in other vaccine developments, this parameter tends to increase because of the presence of whole cells of B. pertussis in formulations that do not have a high degree of purification. Therefore, there are residues from the fermentation process and substances from bacterial metabolism itself that may increase the pH. Nonetheless, the use of the 4 mM PBS buffer solution is able to control this parameter during the storage period of 36 months, thereby ensuring a value close to neutrality. Another relevant aspect is that this 4 mM PBS buffer solution, when added to the vaccine, ensures that osmolarity range of 280-300 mOsmol/kg, which is appropriate for a parenteral product.
The pH of an injectable product is one of the parameters producing adverse events such as pain, burning, reddening and inflammation after a vaccine is administered [42].
Thimerosal content
The Ecuadorian pentavalent vaccine is presented in a multidose form, i.e. in a 6R vial that includes 10 children’s doses of 0.5 mL each. A preservative is recommended for this type of presentation in order to avoid contamination through microorganisms during the extraction process of the different vaccine doses.
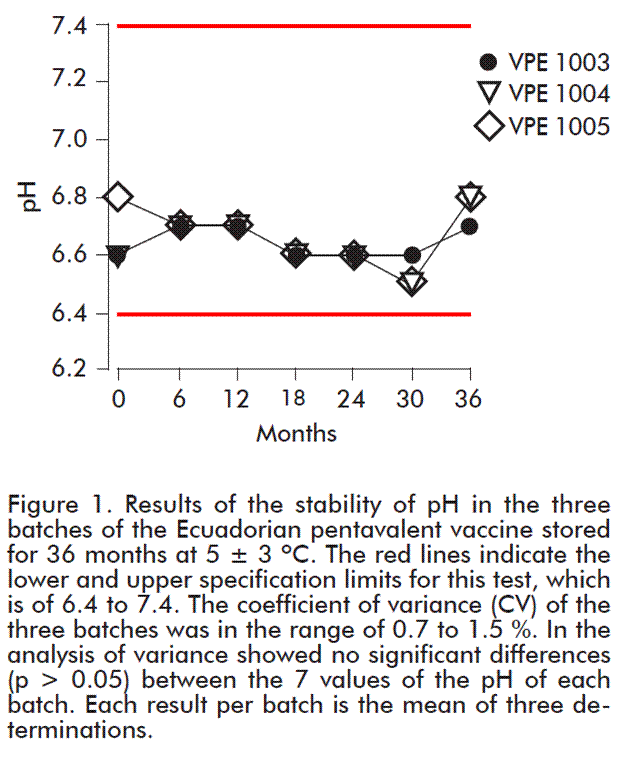
Figure 2 shows the behavior of the thimerosal content in the three batches stored for 36 months at 5 ± 3 ºC. The results of the thimerosal content for the 3 batches reached values ranging 0.010-0.013 g%, which complied with the 0.005-0.02 g% specification, representing a positive result in the determination of the effectiveness of the preservative.
The variability of the test was of 8.2-9.5 %, within the 10 % variation limits. The values reported throughout the study for each batch do not show significant differences (p > 0.05) with a confidence level of 95 %. In the determination at 36 months of stability of each batch, the thimerosal content showed values very similar to the initial ones, which is very important in the course of the useful life of a vaccine.
One of the advantages of combined vaccines is that they reduce the thimerosal content because in monovalent vaccines of hepatitis B, H. influenzae type b, and the combined DPT vaccine, this preservative is applied in each dose independently, but when using the combined DPT-HB-Hib pentavalent vaccine, thimerosal content received by the child is decreased in 75 %. This is very important since certain national Public Health authorities are trying to replace the vaccines containing thimerosal as a precaution measure, although for a number of years WHO has expressed that there is no sign of toxicity derived from the mercury contained in the vaccines [43].
Aluminum ion content
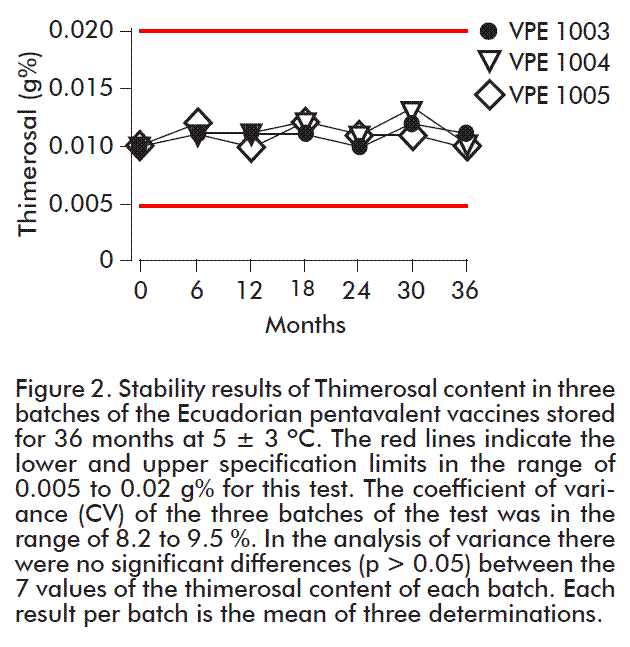
The aluminum ion content values of the Ecuadorian pentavalent vaccine are shown in Figure 3. Results were obtained in the range 0.30-0.39 mg Al3+/mL of the vaccine, with 4.3-9.1 % of variation, thereby complying with the parameter that should be below 10 %. The values reported throughout the study for each batch did not show significant differences (p > 0.05) for a confidence level of 95 %.
It is important to point out that, during this development stage, the quality specification of the pentavalent vaccine is less or equal to 1.25 mg Al3+ per vaccine dose. This was established from the historical data of the production of the Ecuadorian triple DPT vaccine. It was defined, however, that a lower value should be established after a larger number of productive batches would be available. This is a consequence of the fact that according to the results of the evaluation of potency and immunogenicity of the vaccine antigens, as well as the toxicology studies, it was shown that a lower content of this ion does not affect the quality of the vaccine, although the value of 1.25 mg Al3+ per dose has been recommended by FDA. There are, however, national regulatory agencies that consider this value too high and propose a reduction in the specification of this parameter, where even WHO proposes values below 0.85 mg Al3+ per dose. Nevertheless, in the design of the Ecuadorian pentavalent vaccine, the value of this parameter is lower than the maximum recommended by both agencies. This is because adverse reactions at the injection site were shown to be produced by products containing aluminum, which include induration, subcutaneous nodes, inflammation and erythema [44].
Another element contributing to the reduction of this parameter is the combination of this pentavalent DPT-HB-Hib vaccine that reduces in 75 % the aluminum ion content compared to the separate DPT, hepatitis B and H. influenzae type b vaccines.
One important aspect to maintain quality aluminum adjuvant is the storage temperature, is known, that the DPT-HB-Hib pentavalent vaccines should not be stored at above 10 ºC or under 0 ºC, since these conditions affect the quality of the adjuvant and vaccine, also according to the guidelines, the validity period of the product is defined by the true stability study.
Toxicological tests
General safety
The results of the general safety study of the three batches of the Ecuadorian DPT-HB-Hib vaccine demonstrated that the vaccine is not toxic, since the animals did not show signs of toxicity, altered behavior, according to the daily clinical observations, or deaths after up to 36 months of storage of the batches of the vaccine.
In the design of the formulation, different components were used (antigens, adjuvants, diluents and preservatives) that can, singly or combined, produce adverse events. Nevertheless, their quality and concentrations were carefully selected to avoid or minimize these possible events once administering the vaccine preparation.
Specific toxicity of anatoxins and B. pertussis
The results of this assay during the stability study of the 3 batches demonstrated that the animals did not show clinical symptoms on the days following the inoculations. On analyzing live weight gain, compared to the general statistics, no statistically significant differences were observed (p > 0.05) between the experimental groups after applying the vaccines. All groups showed live weight gains after 6 weeks for the analysis of the diphtherial and 4 weeks for the tetanus anatoxins. No typical symptoms of diphtheria and tetanus were observed in the guinea pigs.
One of the most widely used tests to evaluate residual toxicity of the pertussis component is the weight gain test in mice. It is known that mice are not the appropriate biomodels for the clinical study of whooping cough, but intraperitoneal inoculation produces their death due to toxemia [45]. The presence of residual toxins induces significant disorders in live weight gains in the appropriate lines of mice.
In the case of B. pertussis after 7 days, the average weight gain per mouse was of over 60 % compared to the control group. At the end of the study, no mice receiving the batches been analyzed had died.
These results show that the method for the detoxification of diphtherial and tetanus anatoxins, as well as the procedure for the inactivation of the whole cells of B. pertussis, were effective. On the other hand, the formulation technology used to manufacture the vaccine did not produce reversion to toxicity of the anatoxins, which was demonstrated in the 3 batches stored up to 36 months.
Sterility
No contamination was observed in any of the lots evaluated during the stability study. This indicates that the material and solution preparation process, the formulation procedure, the areas involved, the bottling system and sealing of the vaccine preparation, as well as the concentration and type of preservative, are able to ensure the sterility of the product.
Identity tests
Identity of the diphtherial and tetanus anatoxins, Bordetella pertussis, HBsAg and PRP-T
The five antigens forming part of the Ecuadorian pentavalent vaccine were identified in the 3 study batches.
The identification of these antigens throughout the study is able to predict the possibility that their protective capacity will be maintained after their inoculation. The potency tests, however, complement these results since in some cases the response in animals is correlated to their protection in human beings.
Biological tests
Potency of the diphtherial anatoxin
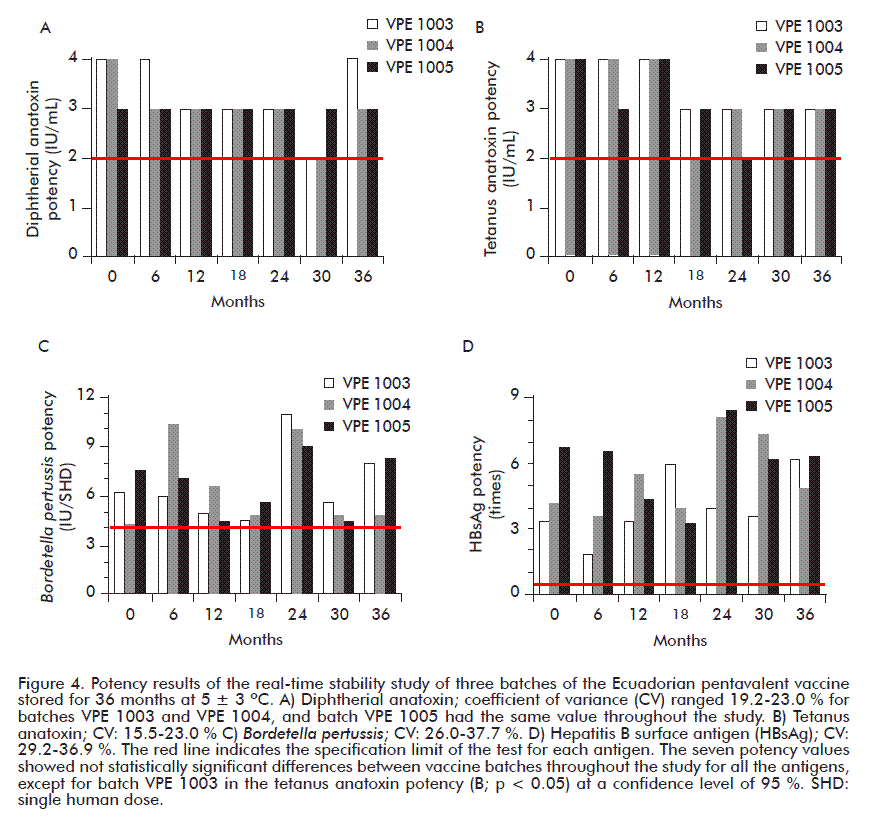
The results of the potency of the diphtherial anatoxin up until 36 months of storage of the 3 batches of the pentavalent DPT-HB-Hib vaccine were equal or better than the specification established of 2.0 IU/mL as observed in Figure 4A.
It is evident that the technology used in the production of the pentavalent vaccine does not affect the biological activity of this antigen. The variability of the results of two batches during the 7 evaluation points was in the range 19.2-23.0 %, which is intrinsic to the test method considering the use of biological systems.
The most conservative reports for biological test variability with animals indicate that this parameter may be above 50 % [8]. No significant statistical differences (p > 0.05) were found in the values reported in batches 1 and 2 of the pentavalent vaccine. Batch 3 was not analyzed because all the results obtained during the study were identical.
Potency of the tetanus anatoxin
The results of the potency of the tetanus anatoxin of up to 36 months of storage in the 3 batches of the pentavalent DPT-HB-Hib vaccine were equal or higher than the specification established of 2.0 IU/mL as observed in Figure 4B.
The technology used in the production of the pentavalent vaccine does not affect the biological activity of up to 36 months of this antigen, however, the statistical analysis showed that the results of the potency of the tetanus anatoxin of batch VPE 1001 was significantly different (p < 0.05) with a confidence level of 95 %. This is because the batch shows a decreasing trend with time since the results are still at a value of 4 IU/mL up until 12 months and later decrease up to 3 IU/mL and remain so until 36 months.
This behavior is not so for batches 2 and 3 where the results showed no significant differences (p > 0.05). These batches showed a decrease of values with time, compared to the initial value, but the values increase at the end of the evaluation.
The variability of the results ranged 15.5-23.0 %, which was below that reported by WHO [8]. WHO has pointed out the problem of the variability of the different tests made in vaccine stability studies, reporting for the potency tests of the diphtherial and tetanus anatoxin a variability of between 50 and 200 %.
Potency of B. pertussis
Figure 4C shows the results of the 3 batches evaluated up to 36 months of storage at 5 ± 3 ºC. All results comply with the quality specification of this parameter, which equals or is higher than 4 IU/SHD.
When analyzing the mean of the results of the 7 sampling points per batch and comparing them, the variability is not evident since the values for batches VPE 1003, 1004 and 1005 were of 6.5 IU/SHD, 6.3 IU/ SHDand 6.5 IU/SHD respectively, nonetheless, the variability was of 26.0-37.7 %. These values are below some reported for this type of test, which is one of the most complicated ones, since it is very inexact and frequently leads to errors that make the tests invalid or rejected and they must then be repeated because of their non-compliance with some validity criterion. This is mainly due to the use of biological systems (quality and type of animal used) and the test method employed (intracerebral challenge), although it must meet all specifications for it to be valid [46].
No significant differences (p > 0.05) were observed between the potency values of B. pertussis for each batch. The potency of B. pertussis has always been a reason for its analysis and discussions in published papers, congresses and technical meetings worldwide. Since the intracerebral method was established for the evaluation of the potency of B. pertussis, the vaccines produced and submitted to this conformity test with the above mentioned criterion have been used by different companies in the world without any risk in human beings. It is known, however, that there are no well characterized immunological indicators of the protective effect of the whole cell pertussis antigen in human beings.
In a meeting held in Geneva, Switzerland, by the working group of WHO on July 20-21 of 2006 and March 28-30 of 2007 with the aim of reviewing the Laboratory Manual for quality control of DPT vaccines [47], the experts agreed that this intracerebral challenge method is still employed for the control of these vaccines although it is known that there is a great deal of variability in this test because companies use different strains, animals of the same or of different sexes, and other differences, which are still a problem today.
Relative in vivo potency of HBsAg
The results of the 3 batches evaluated for 36 months were satisfactory, since they met the specification of 0.5, and they even reached a range of 1.8 to 8.5 times higher than the potency value of the reference material as observed in Figure 4D.
The reference material is a monovalent recombinant hepatitis B vaccine, but the HBsAg component in the Ecuadorian pentavalent vaccine is combined with other antigens where a potentiation effect of the biological response of HBsAg is expressed; this is mainly due to the presence of other antigens in the formulation, acting as adjuvants, as in the case of B. pertussis, which has structural elements in the bacteria such as the lipopolysaccharides that have immunomodulating properties [48].
This result has also been expressed in the development of other combined vaccines, for example in the tetravalent Trivac HB® (DPT-HB) vaccine, the potency of the HBsAg in several batches was higher than the control batch because of the presence of the B. pertussis in the formulation [46].
The variability of the results ranged 29.2-36.9 %, in agreement with previous reports and to the above mentioned discussion for biological tests [49]; some variability reports of the potency test in vivo of the hepatitis B vaccine can even reach a range of 33-300 %, which influences the analyses of the final results of a stability study [8]. No significant differences (p > 0.05) were found in the values of the potency of the HBsAg in the analysis of each batch during the study.
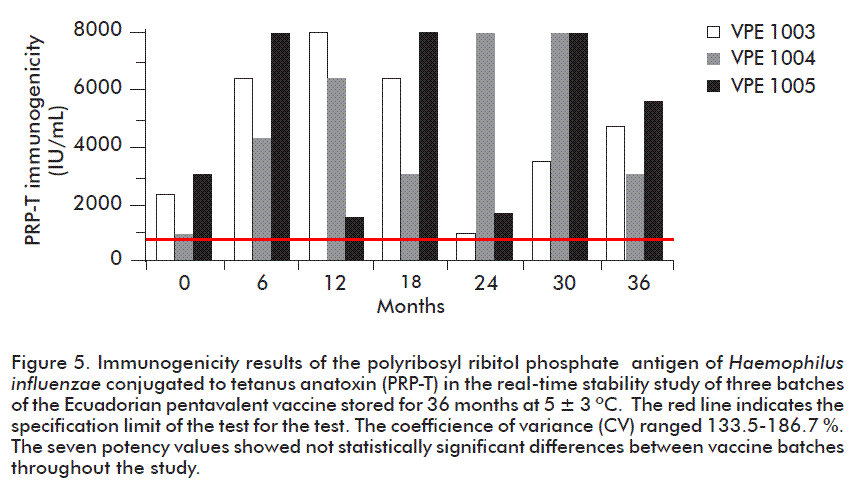
Immunogenicity of the PRP-T component
The results of immunogenicity of the PRP-T components in the 3 batches were high, as observed in Figure 5. This antigen was therefore found to be very immunogenic since 100 % of the animals inoculated showed seroconversion at the 7 points of the sample in each batch until the end of the study at 36 months. The titers reached at all times were of over 1600 IU/ mL, which represents twice the approval limit, except during the release of batch VPE 1004 with the low-est result of 918 IU/mL and batch VPE 1003 at 24 months with 1055 IU/mL, although in both cases they met the specification limit.
In experimental immunogenicity experiments in our laboratory we have obtained anti-PRP-T antibody titers in combined vaccines that are higher than the monovalent Hib vaccine. For example in bivalent vaccines of HB-Hib the anti–PRP-T results were 6 to 12 times those obtained with the monovalent Hib vaccine. This same result was found in pentavalent vaccines, so that it is believed that HBsAg and perhaps B. pertussis favor this immunological potentiation process of the antibody response to Hib.
Another aspect expressed in our results was the high variability found, with a range of 133.5- 186.7 % although this was lower than the coefficient of variability for this test during the validation process which had a value of about 300 %; this, nonetheless, demonstrated that the test can be used as an indicator of stability. No significant differences (p > 0.05) were found in the results obtained for each batch.
CONCLUSIONS
Very few companies have been able to obtain a liquid pentavalent vaccine with its five antigens mixed in a single formulation. Only one of these vaccines has achieved this status in Latin America, i.e. Heberpenta®-L, produced by the CIGB in Cuba.
ENFARMA EP of Ecuador is working in a cooperation project with the CIGB to evaluate its own pentavalent vaccine in later stages, since the current results show that the Ecuadorian pentavalent vaccine is stable when stored up to 36 months at 5 ± 3 ºC. At this temperature, the physicochemical and biological parameters meet all specifications established in each one of the three batches repeatedly in the 7 sampling points, as of their release and up to 3 years at the end of the study.
Physicochemical results were very stable; the pH remained at a range of 6.6 to 6.8 for 36 months, and the same occurred for the thimerosal and the aluminum ion. The behavior of these parameters is fundamental for the stability of the vaccine and for achieving a product with a toxicological profile, as demonstrated in the general safety results.
The antigens were identified throughout the study. These results were endorsed by those obtained in biological trials where they all fulfilled the established specifications, and although variability was found it was lower than that reported for these trials. There were no significant differences between the results obtained for the batches in any of the cases with the exception of the tetanus anatoxin potency, found in only one batch where there the value tended to decrease with time, although the final value was higher than the specification limit.
Altogether, these studies demonstrate that the technology used for the formulation of the Ecuadorian pentavalent vaccine, as well as the excipients and concentrations employed, offer the necessary conditions for long-term stability, since none of the immunological properties of any of the antigens involved in the vaccine are affected. Hence, the vaccine is ready for its evaluation in non-clinical and clinical trials in human beings.
REFERENCES
1. Galazka AM. Stability of vaccines. Expanded Programme on Immunization. WHO, EPI/GEN/89.8; Geneva: WHO; 1989.
2. Requirements for diphtheria, tetanus, pertussis and combined vaccines. TRS 800; 1990.
3. WHO. Regulation and licensing of biologicals products in countries with new developing regulatory authority. Geneva: WHO Technical Report Series No. 858; 1994. p. 3-4.
4. Comité de Expertos de la OMS en especificaciones para las preparaciones farmacéuticas. Directrices para las pruebas de estabilidad de productos farmacéuticos que contienen sustancias medicamentosas bien establecidas en formas farmacéuticas corrientes. In: OMS. Comité de Expertos de la OMS en especificaciones para las preparaciones farmacéuticas, editors. 34° Informe. Serie de Informes Técnicos; No. 863. Ginebra: Organización Mundial de la Salud; 2000. p. 71-85.
5. Chen RT, Shimabukuro TT, Martin DB, Zuber PLF, Weibel DM, Sturkenboom M. Enhancing vaccine safety capacity globally: A lifecycle perspective. Vaccine. 2015;33:D46-D54.
6. Debesa GF, Fernández AR, Pérez PJ. La caducidad de los medicamentos: justificación de una duda. Rev Cubana Farm. 2004 [cited 2015 Oct 5];38(3). Available from: http://scielo.sld.cu/scielo.php?script=sci_ arttext & pid=S0034-75152004000300010
7. WHO. Requirements for MMR and combined vaccine (live). TRS 840. Annex 3. Geneva: WHO; 1992.
8. Jadhav SS, Dogar V, Gautam M, Gairola S. Stability testing of vaccines: Developing Countries Vaccine Manufacturers’ Network (DCVMN) perspective. Biologicals. 2009;37(6):360-3.
9. Pan American Health Organization. Thermostability of Vaccines. Pan Am J Public Health. 1999;6(2):139-41.
10. Ricote-Lobera I, Ortiz-Martín B, Fraile-Gil S, Santos-Mena B, Hidalgo-Correas FJ, García-Díaz B. Estabilidad de los medicamentos termolábiles ante una interrupción accidental de la cadena de frío. Farm Hosp. 2014;38(3):169-92.
11. Andre FE. Development of combined vaccines: manufacturers’ viewpoint. Biologicals. 1994;22(4):317-21.
12. Corbel MJ. Control testing of combined vaccines: a consideration of potential problems and approaches. Biologicals. 1994;22(4):353-60.
13. Vose JR. Pharmaceutical aspects of combined vaccines: a manufacturer’s perspective. Biologicals. 1994;22(4):335-8.
14. Ada G. Combination vaccines: present practices and future possibilities. Biologicals. 1994;22(4):329-31.
15. Redhead K, Sesardic D, Yost SE, Attwell AM, Watkins J, Hoy CS, et al. Combination of DTP and Haemophilus influenzae type b conjugate vaccines can affect laboratory evaluation of potency and immunogenicity. Biologicals. 1994;22(4):339-45.
16. Fenyves A. Regulatory practices and problems with vaccine stability studies and the establishment of shelf lives. Dev Biol Stand. 1996;87:329-33.
17. International conference on harmonization. ICH Topic Q6B: Specifications: test procedures and acceptance criteria for biotechnological/biological products. Step 4. Geneva: ICH; 1999.
18. Ellis RW. Development of combination vaccines. Vaccine. 1999;17(13-14): 1635-42.
19. Expósito N, Martínez E, Alvarez G, Riera V, Proaño H, García S, et al. Pre-formulation study of a pentavalent DTP-HB-Hib vaccine obtained in Ecuador. Biotecnol Apl. 2015;32(4):4251-61.
20. International conference on harmonization. ICH Harmonized tripartite Guideline Stability testing of new drug substances and products Q1A(R2). Geneva: ICH;2003.
21. British Pharmacopoeia. London: Her Majesty’s Stationery Office; 2004.
22. ENFARMA EP. POE 04.019. Procedimiento para la inspección visual las vacunas envasadas y etiquetadas; Quito: ENFARMA EP; 2014.
23. United States Pharmacopoeia 30. National Formulary 25. United States Pharmacopoeia Convention; Inc.: Rockville, MD; 2007.
24. ENFARMA EP. POE 01.001. Procedimiento para la medición del pH en vacunas. Quito: ENFARMA EP; 2014.
25. ENFARMA EP. POE 01.005. Procedimiento para la determinación del tiomersal en vacunas. Quito: ENFARMA EP; 2014.
26. ENFARMA EP. POE 01.023. Procedimiento para la medición del contenido de ión aluminio en vacunas. Quito: ENFARMA EP; 2014.
27. ENFARMA EP. POE 01.012. Procedimiento para la determinación de la seguridad general en vacunas. Quito: ENFARMA EP; 2014.
28. ENFARMA EP. POE 01.014. Procedimiento para la determinación de la toxicidad específica de las anatoxinas diftérica y tetánica en vacunas. Quito: ENFARMA EP; 2014.
29. ENFARMA EP. POE 01.011. Procedimiento para la determinación de la toxicidad específica de la Bordetella pertussis (test de ganancia en peso) en vacunas. Quito: ENFARMA EP; 2014.
30. ENFARMA EP. POE 01.020. Procedimiento para la determinación de la esterilidad en vacunas. Quito: ENFARMA EP; 2014.
31. Ramon G. Floculation dans un élange neutre de toxin-antitoxine diphtériques. CR Soc Biol. 1922;86:6.
32. World Health Organization. Manual for the Production and Control of Vaccines: Tetanus Toxoid. Geneva: WHO; 1978.
33. González P, Martínez S, Ramírez JC, Domínguez F, Díaz Y, Ramírez U, et al. Vacuna antidiftérica-tetánica y antidiftérica-tetánica-pertussis: evaluación del producto final. VacciMonitor. 1999;8(2):8-12.
34. CIGB. PPO.4.09.093.04. Procedimiento para la determinación de la identidad del HBsAg utilizando el sistema ELISA en vacunas. Havana: CIGB; 2013.
35. CIGB. PPO.4.09.041.03. Procedimiento para la determinación de la identidad del PRP-T utilizando el método de inmunoidentificación por Latex en vacunas. Havana: CIGB; 2008.
36. ENFARMA EP. POE 01.021. Procedimiento para la determinación de la potencia de la anatoxina diftérica y tetánica en vacunas. Quito: ENFARMA EP; 2014.
37. Finney DJ. Probit Analysis. 3rd edition. New York: Cambridge University Press; 1971.
38. ENFARMA EP. POE 01.024. Procedimiento para la determinación de la potencia de la Bordetella pertussis en vacunas. Quito: ENFARMA EP; 2014.
39. WHO. Requirement for Hepatitis-B vaccine made by recombinant DNA techniques. TRS 786 Annex 2. Geneva: WHO; 1988.
40. CIGB. PPO 4.09.060.92. Edición 09. Procedimiento establecido para la determinación de la potencia in vivo de la vacuna la hepatitis B recombinante y vacunas combinadas que contengan como componente activo el HBsAg. Havana: CIGB; 2008.
41. CIGB. PPO 4.09.037.03. Procedimiento para la determinación de la inmunogenicidad del componente PRP-T en vacunas. Havana: CIGB; 2013.
42. Ipp M, Cohen E, Goldbach M, Macarthur C. Effect of choice of measles-mumps-rubella vaccine on immediate vaccination pain in infants. Arch Pediatr Adolesc Med. 2004;158(4):323-6.
43. OMS. Inmunización, Vacunas y Productos Biológicos. Tiomersal. Octubre de 2011. 2011 [cited 2015 Oct 5]. Available from: http://www.who.int/immunization/newsroom/thiomersal_information_sheet/es/
44. Jefferson T, Rudin M, Di Pietrantonj C. Adverse events after immunisation with aluminium-containing DTP vaccines: systematic review of the evidence. Lancet Infect Dis. 2004;4(2):84-90.
45. Pittman M. Influence of preservatives, of heat, and of irradiation on mouse protective activity and detoxification of pertussis vaccines. J Immunol. 1952;69:201-16.
46. Expósito NS, Cardoso D, Martínez E, Herrera Y, Cosme K, Díaz PA, et al. Vacuna combinada cubana Trivac HB®. Biotecnol Apl. 2006;23(2):158-64.
47. WHO Working Group meetings on revision of the Manual of Laboratory Methods for testing DTP vaccines. Meeting Report Geneva, Switzerland, 20-21 July 2006 and 28-30 March 2007. 2007 [cited 2015 Oct 5]. Available from: http://www.who.int/ biologicals/areas/vaccines/dtp/DTP_Fi-nal_Combined_Reports_17_12_2007.pdf
48. Chaby R, Caroff M. Lipopolysaccharides of Bordetella pertussis endotoxin. In: Wardlaw A C, Parton P, editors. Pathogenesis and immunity in Pertussis. New York: John Wiley & Sons; 1988. p. 247-71.
49. Organización Mundial de la Salud, (OMS). Guía de la OMS sobre los requisitos de las prácticas adecuadas de fabricación (PAF). Segunda parte: Validación. WHO/ VSQ/97.02. Ginebra: WHO; 1998.
Received in January, 2016.
Accepted in June, 2016.
Néstor Expósito. Dirección de Desarrollo Tecnológico, Centro de Ingeniería Genética y Biotecnología, CIGB. Ave. 31 e/ 158 y 190, Cubanacán, Playa, CP 11600, La Habana, Cuba. E-mail: nestor.exposito@cigb.edu.cu.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}