










My SciELO
 Custom services
Custom servicesServices on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkBiotecnología Aplicada
On-line version ISSN 1027-2852
Biotecnol Apl vol.34 no.1 La Habana Jan.-Mar. 2017
TECHNIQUE
Cloning, expression and purification of the multiepitopic polypeptide TAB1-His for HIV 1 diagnosis
Clonaje, expresión y purificación del polipéptido multiepitópico TAB1-His para el diagnóstico del VIH-1
Yeosvany Cabrera-Artiles1, Isi Veitia1, María T Barceló1, Duniesky Martínez1, Idialis Hernández2, Vladimir Leal1, Raúl Armas1, Julio Alfonso Rubí1
1 Laboratorio de Biología Molecular, Centro de Ingeniería Genética y Biotecnología de Sancti Spíritus. Apartado 83, Sancti Spíritus, CP 60200, Cuba.
2 Laboratorio de Retrovirus, Centro de InmunoEnsayo, La Habana, Cuba.
ABSTRACT
The multi-epitope polypeptide TAB1 representing sequences of the gp120 HIV protein is one of the antigens included in the UMELISA® HIV 1+2 RECOMBINANT diagnostic kit to detect antibodies against HIV. This test is produced by the Immunoassay Center and it has been used in Cuba both for screening HIV infected patients and checking blood quality in blood Banks. Originally the protein was expressed in a vector with the tryptophan promoter and was purified by reverse phase HPLC, which is an expensive and low yielding process. In this study, the expression vector was redesigned, using new cloning and purification techniques, by including a gene modification that added a hexahistidine tag to TAB1, allowing for its purification with immobilized metal affinity chromatography. The replacement of the tryptophan promoter regulatory system by the hybrid ptrc promoter offered better control and an expression level of about 20 %. This new process enabled the production of the TAB1-His protein with the appropriate purity profile and antigenic properties for its binding to HIV antibodies, while meeting the requirements for its use in the diagnostic kit UMELISA® HIV 1+2 RECOMBINANT.
Keywords: HIV, gp120, IMAC, recombinant protein, multiepitopic polypeptide.
RESUMEN
El polipéptido multiepitópico TAB1, que representa regiones de la proteína gp120 del Virus de la Inmunodeficiencia Humana (VIH), forma parte de los antígenos que componen el juego diagnóstico UMELISA® HIV 1+2 RECOMBINANT para la detección de anticuerpos anti-VIH. Este inmunodiagnóstico producido por el Centro de Inmunoensayo (CIE), es el que se utiliza en Cuba, tanto para la pesquisa de pacientes infectados con el VIH como para asegurar que toda la sangre que reciben los bancos esté libre del virus. Originalmente la proteína se expresaba en un vector bajo el promotor triptófano y su purificación se realizaba por HPLC de fase reversa, lo que encarecía el proceso y se obtenían bajos rendimientos. En este trabajo, con el empleo de nuevas técnicas de clonaje y purificación se realizó un rediseño del vector de expresión, que incluyó la modificación del gen para incorporarle una cola de histidinas a la proteína TAB1, lo que permitió purificarla por cromatografía de afinidad a iones metálicos inmovilizados. La modificación del sistema regulador del promotor triptófano por el ptrc ofreció un mejor control y niveles de expresión alrededor del 20 %. Con este nuevo proceso productivo se obtuvo una proteína TAB1-His con un grado de pureza y propiedades antigénicas que permiten utilizarla para la detección de anticuerpos anti-VIH y, por tanto, cumple con los requisitos exigidos para su empleo en el diagnosticador UMELISA® HIV 1+2 RECOMBINANT.
Palabras clave: VIH, gp120, quelatos metálicos, proteína recombinante, polipéptido multiepitópico.
INTRODUCTION
The Human Immunodeficiency Virus (HIV) epidemic is one of the greatest problems in public health, after it has produced more than 34 million deaths. The World Health Organization (WHO) reported that at the end of 2014 there were 36.9 million persons infected with HIV worldwide [1].
In HIV infected patients, the symptoms and clinical signs are non-specific and they vary greatly from one individual to another, and most patients may even be completely asymptomatic. All persons infected, however, develop antibodies against the virus proteins. These antibodies circulate through the blood of patients and can be detected by laboratory methods that are the key to its diagnosis [2, 3]. Generally, they are based on the techniques of the enzyme-linked immunosorbent assay (ELISA) making it possible to process a large number of samples at the same time. Its high sensitivity ensures the safety of the blood and its derivatives [4, 5].
Most modern methods use ELISA plates coated with a combination of recombinant proteins and synthetic peptides that represent different immunogenetic regions of the main structural proteins of the virus [6-8]. The ELISA test used in our country for the epidemiological screening of the high risk population and the control of the blood and its derivatives, is the UMELISA® HIV 1+2 RECOMBINANT, based on the SUMA® technology that was developed and produced by the Immunoassay Center (CIE). The combination of antigens used in coating the plates, makes it possible to detect antibodies against HIV-1 and HIV-2 [9, 10]. This diagnostic assay has been essential for the recognition made by the World Health Organization and the Pan American Health Organization in June 2015, where they announced that Cuba had become the first country in virtually eliminating the mother-to-child transmission of HIV [11].
One of the recombinant proteins forming the combination of antigens of the UMELISA® HIV 1+2 RECOMBINANT is TAB1, a multi-epitope polypeptide that was designed at the Center for Genetic Engineering and Biotechnology (CIGB). It contains several epitopes of the gp120 protein that includes the sequence of several viral isolates, thereby detecting the presence of antibodies against gp120 which correspond to different HIV genetic variants. The chimeric gene coding for TAB1 is expressed in Escherichia coli under the regulation of the tryptophan promoter. Once solubilized, the recombinant protein is purified by reverse phase HPLC [12]. Although conventional purification methods cannot be ignored, the inclusion of affinity tags has become the standard for the production of recombinant proteins since they enable their recovery and purification at a low cost. The most popular fusion is that including poly-histidine tags (His-tag) because they can also be used under native or denaturalized conditions [13]. This paper reports the inclusion of a histidine tag to the TAB1 protein and its purification under denaturalizing conditions in a single immobilized metal ion affinity chromatography (IMAC) step using the Ni-NTA Agarose matrix.
MATERIALS AND METHODS
Biological materials and chemical substances used
The restriction and modification enzymes were obtained from Heber Biotec S.A. (Cuba), New England Biolabs (USA) and Promega (USA), and specifications from suppliers were followed for their use. The reagents, culture media components and materials employed were supplied by Merck (Germany), Oxoid (England), Sigma-Aldrich (USA), Biorad (USA), GE HealthCare (USA), and Biocen (Cuba). DNA sequencing was made by contracting the automatic sequencing service from Macrogen (Korea).
Bacterial strain and culture media
The bacterial strain Escherichia coli XL-1-Blue [thi rec A1 endA1 gyrA96 (Nalr) hsdR17 (rk- mk+) supE44 relA1Δ (lac-proAB) F’ proAB LacIq Z DM15 Tn10] was used as host for recombinant plasmids carrying the TAB1-coding gene [14]. All cultures were made in the Luria-Bertani (LB) medium containing 5 g/L yeast extract, 10 g/L tryptone, 10 g/L NaCl, pH 7.5, supplemented with 100 μg/mL ampicillin (LBA). For the solid medium, Technical Agar No.3 was added at 15 g/L.
Plasmid DNA and genes
The following plasmid DNA were used: expression vector pIT120, containing the gene coding for TAB1 (multi-epitope polypeptide formed by peptides from 3 different isolations (MN, SC and WMJII) of the hyper-variable loop V3, formed by 15 amino acids each and a peptide of 11 amino acids of the conserved C1 region of the gp120 of the HIV-1, linked by a stabilizing peptide AGGGA and its fusion with the first 58 amino acids of the N-terminal end of the human interleukin 2) [12]; the pGEM-T Easy vector plasmid (Promega, USA) to clone the products from amplified PCR with Taq DNA Polymerase I; the expression vector pBadMyc His A (Invitrogen, USA) for linking the gene to a hexahistidine tag at the C-terminus end of the protein for its later purification by IMAC, and the pTrcHisA vector containing the ptrc promoter (Invitrogen, USA). The empty pTBad vector was used as negative control.
DNA primers for PCR amplification of the gene coding for the TAB1 protein
To amplify the coding region of the TAB1 protein, the following oligonucleotides were used: sense (25 nucleotides) 5´-ATTCCATGGCGCCTACTTCAAGTTC-3´; anti-sense (22 nucleotides) 5´-CGTCTCGAGTACCATCGTTGCA -3´. The translation start codon ATG translation is highlighted in bold, and restriction sites NcoI (CCATGG) and XhoI (CTCGAG) appear underlined.
Amplification by PCR of the coding region of the TAB1 protein
The amplification of the coding region of the TAB1 protein was carried out with 5 U of Taq DNA Polymerase I, using 5 ng of the pIT120 plasmid as template [12] and 1 μM of each one of the oligonucleotide primers. The reaction was carried out in an Eppendorf Mastercycler Personal thermocycler, where 30 cycles were made comprising one minute of denaturalization at 94 ºC, 30 seconds of hybridization at 55 ºC, and 40 s of extension at 72 ºC in a reaction volume of 50 μL with dNTPs at a final concentration of 200 μM.
Cloning the coding region of TAB1 under the pbad and ptrc promoters
The fragment amplified with Taq pol I, from the region of 443 bp coding for TAB1, was ligated to the pGEM-T Easy cloning vector for PCR fragments. The pGEM-T Easy/TAB1 construction was digested by the endonucleases NcoI/XhoI and the fragment of interest was subcloned under the pbad promoter, linked to the histidine tag and the transcriptional terminator rrnB of vector pBadMyc HisA, previously digested with the NcoI/SalI endonucleases, resulting in the pBadTAB1 construction.
To place the gene under the ptrc promoter, the pBadTAB1 was digested with the NcoI/PvuI endonucleases and subcloned into vector pTrcHis A which had been previously digested with the NcoI/PvuI endonucleases and dephosphorylated. The resulting construction was named pTBadTAB1.
Expression of the TAB1-His protein in E. coli XL-1-Blue cells
The E. coli XL-1-Blue cells transformed with the pBadTAB1 plasmid were cultured in 50 mL of LBA medium and grown at 37 ºC, under agitation at 250 rpm in an orbital shaker INFORS HT (Switzerland). The growth was followed by optical density at 620 nm (OD620 nm). Recombinant protein expression was induced with arabinose at a final concentration of 0.2 % (w/v) when the OD620 nm ranging 0.5-0.7 and the culture was grown for the following 10 h. Samples of culture volumes equivalent to the ratio 2/OD620 nm, were collected, and centrifuged at 12 000 × g for 3 min at 4 ºC.
Cultures were also established for the expression of TAB1-His with the pTBadTAB1 plasmid under the same conditions, and protein expression was induced by adding IPTG (isopropyl-β-D-1-thiogalactopyranoside) to a final concentration of 1 mM, and the incubation was maintained for a further 8 h.
Fermentation of E. coli XL-1-Blue [pTBadTAB1]
The fermentation process was carried out in an INFORS HT bioreactor with a capacity for 7.5 L and a working volume of 5 L. The following parameters were fixed and recorded during the culture process through the Iris v. 5.0 software: stirring speed 500 rpm, air flow 1 vvm, pH 7.2 controlled by NaOH (40 %; w/v) and H3PO4 (40 %; v/v), at 37 ºC. The bioreactor was inoculated with 50 mL of a culture that had been growing in Erlenmeyers with 100 mL of the LBA medium for 14 h in a thermostatic orbital shaker at 37 ºC and 250 rpm. Induction was made with IPTG at different concentrations: 0.25 mM, 0.5 mM and 1 mM. After 15 h, the fermentation was stopped. The resulting biomass was used for the purification process.
Purification of the TAB1-His protein by affinity to Ni-NTA Agarose
For cell disrupture, 15.6 g of biomass were resuspended at a proportion of 1:10 (w/v) in rupture buffer (20 mM Tris-HCl, pH 7.9; 500 mM NaCl). It was homogenized with Polytron Ultra-Turrax T25 IKA® (Germany) at 9500 rpm for 5 min and sonicated 6 times for one minute and one minute standing on ice; for ultrasonication we used the Vibracell Sonics & Materials (USA) with a 6 mm diameter tip at a potency of 35 W and a frequency of 100 Hz. The material resulting from cellular rupture was centrifuged at 20 000 × g for 30 min at 4 ºC; the pellet was collected and the extraction and mixing buffer (20 mM Tris-HCl; pH 7.9; 500 mM NaCl; 5 mM imidazol; 6 M urea) was added at a proportion of 1:5 (w/v). It was homogenized with Polytron Ultra-Turrax T25 at 9500 rpm for 5 min and incubated for 1 h on ice. It was then centrifuged at 20 000 × g for 30 min at 4 ºC and the supernatant from centrifugation was collected by decanting. The sample filtered at 0.2 μm was applied on a XK 26 GE Healthcare column (USA) with a height of 9.5 cm, having a 50 mL matrix of Ni-NTA (nickel-nitrilotriacetic acid) Agarose (QIAGEN, USA), with a binding capacity of 5 mg of protein per mL of gel at a flow of 10 cm/h, which was previously equilibrated with the binding buffer. Washing and elution were carried out in binding buffer with 20 and 300 mM imidazol, respectively.
Protein assay
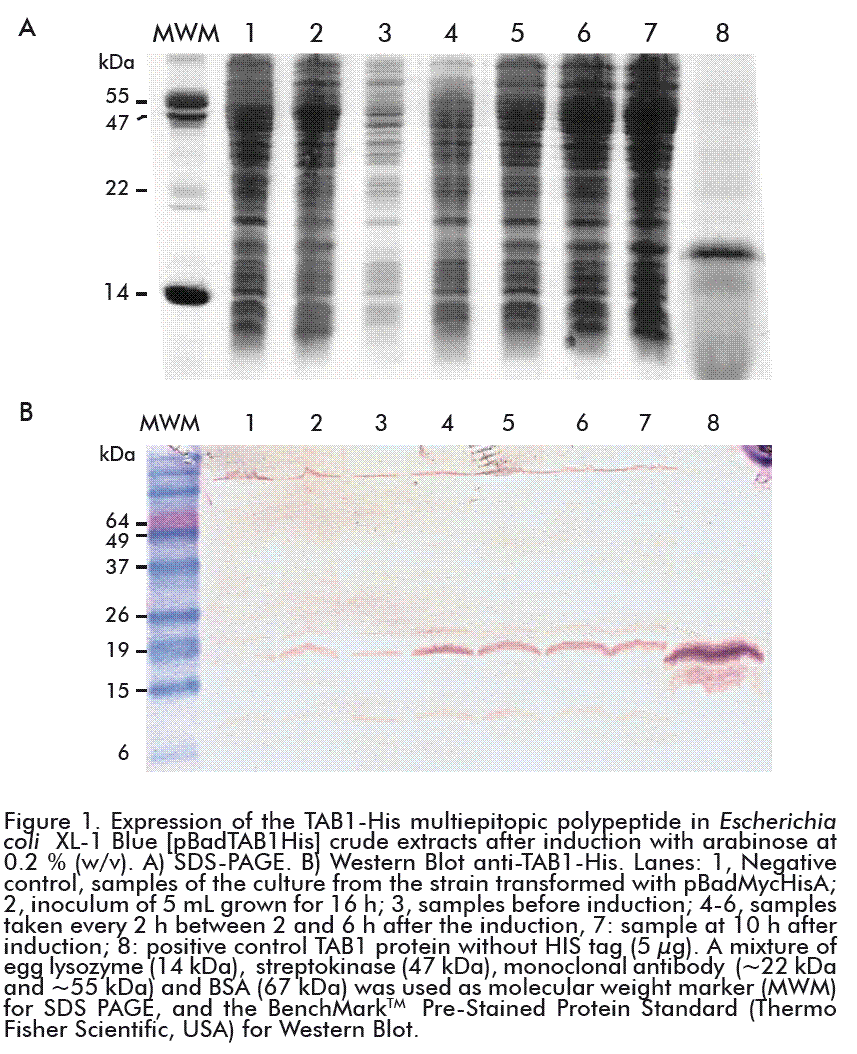
The concentration of proteins was carried out by the BCA method (bicinchoninic acid) using the Thermo Scientific Pierce™ BCA Protein Assay Kit (USA) and using bovine serum albumin (BSA) as the standard [15, 16]. Proteins were separated by SDS-PAGE (15 % polyacrylamide, 2 % SDS) [17]. Samples were diluted in the reducing loading buffer and heated for 10 min at 100 ºC. About 20 μg of total protein per well were applied, or in the case of fractions of the purification, a constant volume of 20 μL was used. The control employed was 15 μg of purified TAB1. For the densitometry estimation of the percentage of the expression and purity, we used the ImageJ 1.44p program [18] on the digitalized image of the protein gel. The Western Blot anti-TAB1-His was made with a pool of the inactivated sera of patients infected with HIV, supplied by the AIDS Research Laboratory, Cuba. It was developed with a human polyclonal anti-IgG antibody obtained from sheep and conjugated with alkaline phosphatase. The insoluble and stained substrate used was Nitroblue Tetrazolium/5-Bromine-4-Chlorine- 3-indolyl Phosphate (NBT/BCIP).
To determine the sensitivity and specificity of UMELISA® HIV 1+2 RECOMBINANT (TecnoSuma®, Cuba) we substituted the TAB1 protein by the purified TAB1-His, in the mixture of antigens used in the coating solution. First we determined the optimum working concentration of the protein of interest that was of 0.5 μg/mL and we later coated the plates using the mixture of antigens, and following all the recommended specifications for the production of the diagnostic kit.
Sensitivity evaluation used four reference serologic panels: Certification Panel of the Quality Assurance and Quality Control Department (DACCA according to its Spanish acronym) of the Immunoassay Center, Havana, Cuba; Functioning Panel Anti-HIV-1/2 Combo PRZ 205, Anti-HIV 1 Panel Mixed Titer PRB 204 and the Seroconversion Panel HIV-1 PRB 931 (Sera Care Life Sciences, USA - BBI Diagnostics). For specificity we used samples of negative sera from blood donors of the Havana Provincial Blood Bank. All samples were considered positive when the ratio (Fm-B)/(P-B) was higher than or equal to 0.27. Where Fm, P and B refer to the fluorescence of the sample evaluated, the positive control concerning the UMELISA® HIV 1+2 RECOMBINANT, and the blank, respectively.
The sensitivity of antibody detection was estimated as the percentage of samples considered to be positive with the TAB1His protein in relation to those considered to be positive with the TAB protein under the conditions described above. Specificity was defined as the percentage of negative HIV samples that were considered negative with the TAB1His protein, compared to those considered negative with the TAB protein in the UMELISA® HIV 1+2 RECOMBINANT kit.
Statistical analysis
In order to assess the effect of the IPTG concentration on protein productivity, a Mann-Whitney U test was applied to IPTG at two concentration levels, with differences considered significant for p < 0.05. Values were expressed as the arithmetic mean ± standard deviation of three replicates. The statistical analysis was made through SPSS (Statistical Package for Social Sciences) version 15.0.
RESULTS AND DISCUSSION
Cloning the gene coding for the TAB1 recombinant protein in the E. coli expression vectors pBadMyc His A and pTrcHis
A 443-bp long fragment of the gene coding for TAB1 was PCR-amplified from the pIT120 plasmid, with primers designed as reported for this chimeric gene [12]. The antisense oligonucleotide introduces an XhoI site and eliminates the stop codon, enabling the translational fusion to the hexahistidine tag at the C-terminus. A His-tag N-terminal fusion could eliminate the expression enhancer effect of the human Interleukin-2 fragment on TAB1.
In fact, Duarte et al. reported that the TAB1 protein increased the levels of expression up to 20 % when the coding gene was linked to the first 58 amino acids of the N-terminal end of the human Interleukin-2. Nevertheless, its expression was not detected when it was cloned directly under the same promoter in the absence of the IL-2 fragment. A plausible explanation is that the IL-2 fragment would be able to confer resistance to the bacterial proteases through the rapid insolubilization of the protein forming inclusion bodies. An additional possibility would be that the IL-2 fragment would confer a more appropriate structure to the messenger RNA, improving the stability of the molecule and facilitating the translation process [12].
The fragment amplified with Taq pol I was inserted into the PCR cloning vector pGEM-T Easy. The positive clones 3, 4 and 5 were checked by sequencing and the presence of mutations in the coding region was ruled out (data not shown). Clone 3 was selected for subsequent steps.
Then, the pGEM-T Easy/TAB1 construction was digested by the endonucleases NcoI and XhoI and the fragment of interest was sub-cloned to the pBadMyc His A vector that had been previously digested with endonucleases NcoI and SalI. This enabled the transcriptional control of the gene by the pbad promoter, inducible with arabinose, and its fusion to a hexahistidine tag at the C-terminal end, with the aim of purifying the protein by metal ion affinity chromatography.
Subsequently, E. coli XL-1-Blue cells were transformed with the pBadTAB1 plasmid and cultured in a shaker to evaluate the expression of the protein of interest. Although the maximum concentration of arabinose (0.2 %; w/v) reported for this promoter was used, the expression levels did not surpass 8 % of the total proteins at 10 h after induction (Figure 1A, lane 7). Both the TAB1 and the TAB1-His proteins were detected by Western Blot using a mixture of HIV-1 positive human sera. The presence of TAB1-His was also observed in the samples before the induction (lanes 2 and 3), as the product of a basal expression of the gene (Figure 1B). The assay evidenced that the addition of the polyhistidine tag did not affect the immunological recognition of the multi-epitope polypeptide. Because of its small size (0.84 kDa) and its repetitive character, the hexa-peptide showed low immunogenicity, being therefore widely used and without interfering with the immune response against the protein of interest, including the HIV antigenic proteins used in the serological diagnosis [19-21].
In previous studies with TAB1, levels of expression of over 20 % were obtained [12, 22]; it was therefore decided that a new construction should be made under a stronger promoter, to try to increase the levels of expression and enable a better regulation of the expression of the gene in bacterial culture.
For that purpose, the pBadTAB1 construction was digested by the endonucleases NcoI and PvuI and the fragment with the gene of interest linked to the histidine tag and the transcriptional terminator rrnB was cloned into the pTrcHis A vector previously digested with endonucleases NcoI/PvuI and dephosphorylated. The construction obtained made it possible to use the ptrc promoter and the untranslated 5´ region of the latter vector, named pTBadTAB1.
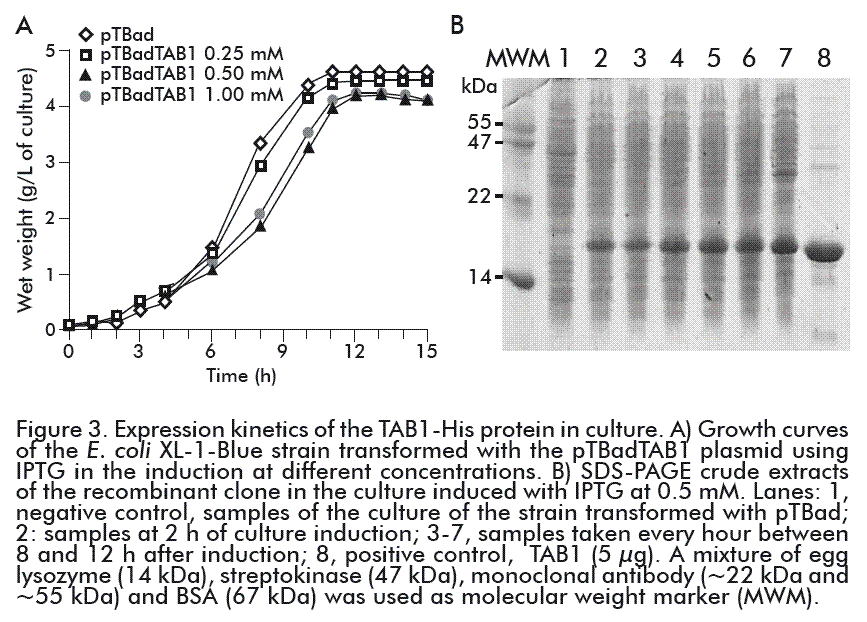
The culture of the E. coli XL-1-Blue strain transformed with the pTBadTAB1 plasmid was induced with IPTG at a final concentration of 1 mM. The TAB1-His protein showed the more intense band among the proteins of the crude bacterial extract; at 8 h after the induction it represented 20 %, according to the densitometric analysis made with the ImageJ 1.44p program. An increase in the level of expression of the recombinant gene in relation to that obtained under the control of the pbad promoter was detected (Figure 2).
Fermentation of E. coli XL-1-Blue transformed with the pTBadTAB1 plasmid
In the 5-L bioreactor, we studied the effect of the concentration of the IPTG inducer in the growth of E. coli XL-1-Blue cells transformed with the pTBadTAB1 construction and the expression of the TAB1-His protein. Figure 3A shows that, when adding IPTG, a decrease in the growth of the strain that contained the gene coding for TAB1-His, compared to the control strain transformed with the pTBad plasmid. This behavior can be attributed to the production of the TAB1-His protein by the bacterium. Regardless of the concentration of IPTG used in all fermentations, the stationary phase was reached at 11 h of culture.
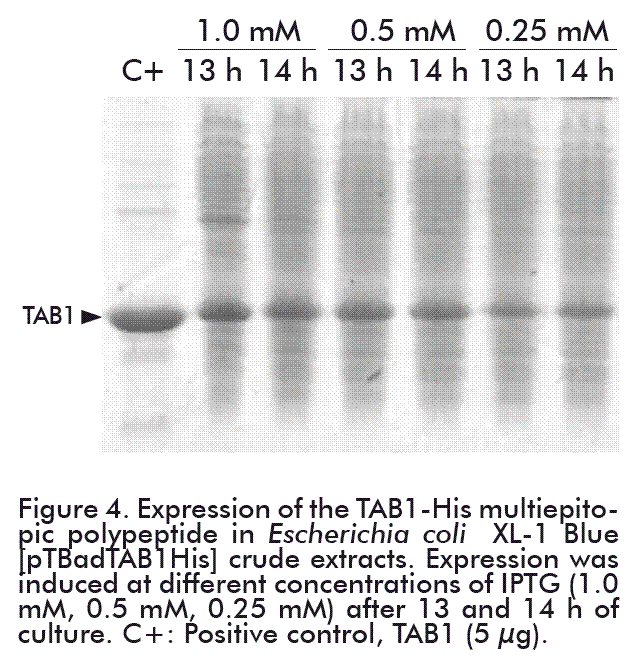
Two hours after induction, the growing expression of the protein of interest became evident. The highest levels were reached as of 10 h after the induction (corresponding to 13 h of culture) (Figure 3B). Nevertheless, the expression levels of the TAB1-His protein in culture (bioreactor) varied on using the different concentrations of the inducer. The levels of TAB1-His protein expression reached at 0.25 mM IPTG were 50 % less than those attained at other inducer concentrations. At 0.5 mM and 1.0 mM IPTG, the levels of expression of the TAB1-His protein were similar to that 20 % TAB1 expression level obtained under the tryptophan promoter (Figure 4) [22].
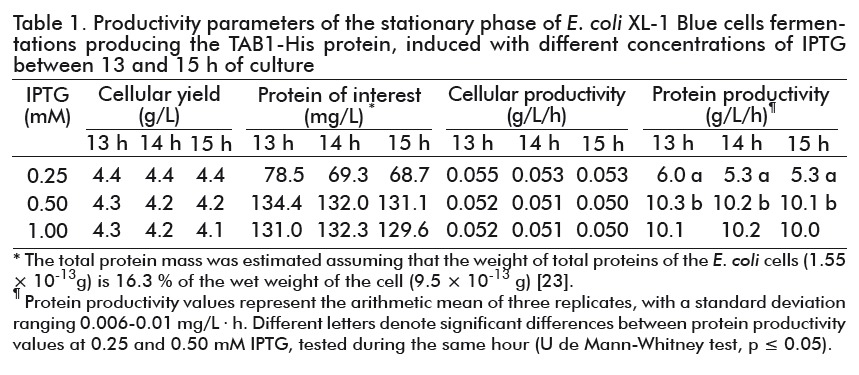
Cellular productivity was similar at the three concentrations of the IPTG used, but greater influence in protein productivity was reached at 0.25 and 0.5 mM IPTG (Table 1). Significant differences were found in this productivity parameter between both concentrations at all time periods studied (Mann-Whitney U test; p = 0.0495). Therefore, 0.5 mM IPTG was chosen as the optimal inducer concentration for the productive process, with the aim of reducing the impact of IPTG high cost and toxicity. As shown in Table 1, there was no variation in cellular or protein productivity at 0.5 mM IPTG between 13 and 15 h of fermentation. Therefore, we decided that the cultures should be carried out for 10 h after the induction.
Purification of the TAB1-His protein by affinity to Ni-NTA Agarose
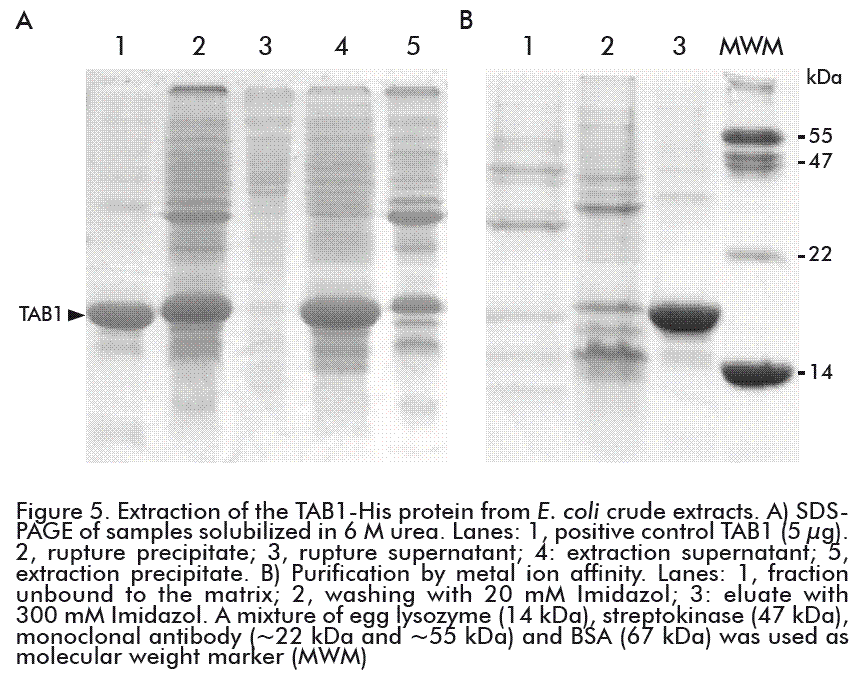
After centrifugation of the cell disrupture extract, the chimeric protein TAB1-His was found in the pellet fraction (Figure 5A, lane 2). This result agrees with previous reports where the TAB1 protein, under the control of the tryptophan promoter, was expressed as inclusion bodies in the cellular cytoplasm [12, 22]. The addition of the hexahistidine to the TAB1 protein did not modified its solubility.
The low solubility of some proteins facilitates the design of a fast and efficient purification procedure.
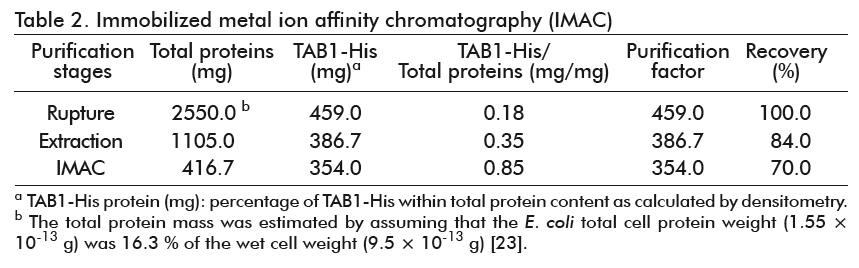
The formation of inclusion bodies composed basically by the protein of interest, can be used as a purification step through a single centrifugation of the insoluble material of the crude extracts and the re-suspension of the protein in urea or guanidium chloride [24]. After the washing steps, the solubilization of TAB1 was made with the use of detergents and 8 M urea [12] or with 8 M guanidium hydrochloride [22]. The extraction of TAB1-His with 6 M urea, directly from the cell disruption pellet, enabled the recovery of 84 % of the protein (Table 2), with about 35 % purity (Figure 5A, lane 4), while preventing the solubilization of the TAB1 aggregated forms and host contaminant proteins (Figure 5A, lane 5).
Under these denaturing conditions, the TAB1-His protein was purified in only one step of metal ion affinity chromatography, with final recovery of 77 %. Through this purification process, we obtained a final sample of TAB1-His protein with more than 85 % purity (Figure 5B, lane 3), which represents a 4.7- fold increase in the purification degree of the initial sample applied to the column. The TAB1 protein was obtained with 95 % purity when it was purified by RP-HPLC for its use as a vaccine antigen [12] and this procedure was followed by Narciandi et al. for obtaining TAB1 for diagnostic purposes [22]. Nevertheless, the production of recombinant proteins by this method involves high cost and lower throughput, representing a disadvantage in relation to purification by IMAC; furthermore, the purity obtained here does not produce any variation in the sensitivity and specificity of the diagnostic kit as explained below.
Evaluation of the sensitivity and specificity of the recombinant protein
A set of HIV positive samples from different reference panels and negative HIV samples from blood donors were used to test the specific recognition and sensitivity of the diagnostic kit UMELISA® HIV 1+2 RECOMBINANT. It was used the TAB1-His purified protein as a part of the antigen mixture in the coating solution and a reference mixture containing the TAB1 protein. The sensitivity of UMELISA with the TAB1-His protein was 100 % in 20 samples of the HIV positive serum and the specificity was of 100 % in the serum samples from the 40 healthy donors. With the DACCA panel, where several samples are found to be in the grey or positivity threshold zone, there was also an agreement of 100 % (Table 3). These results show that the hexahistidine added to the TAB1 protein does not affect its biological activity.
CONCLUSIONS
A new genetic construction was established, which enabled 20 % expression of the TAB1 protein in an insoluble form in E. coli through genetic regulation via the ptrc promoter. The addition of a poly-histidine tag made it possible to carry out the purification of the protein under denaturing conditions by IMAC. The recovery and purity levels obtained in a single chromatographic step shows that it is possible to substitute the reverse phase purification method used previously, without affecting its recognition by the antibodies present in the sera of HIV infected patients. The cloning, expression and purification strategy used here may be applied to other proteins for diagnostic purposes, which ultimately result in a more efficient manufacturing process of the diagnostic kit components.
REFERENCES
1. WHO. Global Health Observatory (GHO) data. Geneva: World Health Organization. 2015 [cited 2016 Dec 17]. Available from: http://www.who.int/gho/hiv/en/
2. Singh AE, Lee B, Fenton J, Preiksaitis J. The INSTI HIV-1/HIV-2 antibody test: a review. Expert Opin Med Diagn. 2013;7(3):299-308.
3. Rayment M, Asboe D, Sullivan AK. HIV testing and management of newly diagnosed HIV. BMJ. 2014;349:g4275.
4. Tiwari RP, Jain A, Khan Z, Kumar P, Bhrigu V, Bisen PS. Designing of novel antigenic peptide cocktail for the detection of antibodies to HIV-1/2 by ELISA. J Immunol Methods. 2013;387(1-2):157-66.
5. Rikhtegaran Tehrani Z, Azadmanesh K, Mostafavi E, Soori S, Azizi M, Khabiri A. Development of an integrase-based ELISA for specific diagnosis of individuals infected with HIV. J Virol Methods. 2015;215-216:61-6.
6. Craske J, Turner A, Abbott R, Collier M, Gunson HH, Lee D, et al. Comparison of false-positive reactions in direct-binding anti-HIV ELISA using cell lysate or recombinant antigens. Vox Sang. 1990;59(3):160-6.
7. Ramos G, Valdespino-Díaz MA, Hernández-Marín M, Valle E, Selles ME, Pozo L. Uso de un péptido sintético de la glucoproteína 36 del VIH 2 en mezclas de antígenos para el inmunodiagnóstico de la infección por VIH tipo 1 y 2. Enferm Infecc Microbiol Clin. 2015;33(10):663-5.
8. Campbell-Yesufu OT, Gandhi RT. Update on human immunodeficiency virus (HIV)-2 infection. Clin Infect Dis. 2011;52(6):780-7.
9. Morales-Rodríguez E, Alonso-RamosV, Pozo-Peña L, González-Suárez A, Figueiras-Lache Y, González-Quintero A, et al. Evaluación de la aplicación del ensayo UMELISA HIV 1+2 RECOMBINANT en muestras de sangre seca sobre papel de filtro. Rev Biomed. 2004;15(3):149-55.
10. Delahanty-Fernández A, Bequer-Ariza DC, Hernández-Marín M, Zulueta-Rodríguez O, Pozo-Peña L, Hernández-Spengler I, et al. Desarrollo de un ensayo sandwich de doble antígeno para la detección de anticuerpos contra el VIH-2 empleando un péptido sintético biotinilado de la proteína gp36. Enferm Infecc Microbiol Clin. 2015;33(7):464-8.
11. Lenzer J. How Cuba eliminated mother-to-child transmission of HIV and syphilis. BMJ. 2016;352:i1619.
12. Duarte CA, Montero M, Seralena A, Valdes R, Jimenez V, Benitez J, et al. Multiepitope polypeptide of the HIV-1 envelope induces neutralizing monoclonal antibodies against V3 loop. AIDS Res Hum Retroviruses. 1994;10(3):235-43.
13. Wingfield PT. Overview of the purification of recombinant proteins. Curr Protoc Protein Sci. 2015;80:6.1.1-35.
14. Bullock WO, Fernandez JM, Short JM. XL-1-Blue: a high efficiency plasmid transforming recA Escherichia coli strains with b-galactosidase selection. BioTechniques. 1987;5:376-9.
15. Smith PK. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150(1):76-85.
16. Wiechelman KJ, Braun RD, Fitzpatrick JD. Investigation of the bicinchoninic acid protein assay: identification of the groups responsible for color formation. Anal Biochem. 1988;175(1):231-7.
17. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680-5.
18. Rasband W. ImageJ. Image processing and analysis in Java. Bethesda: National Institute of Mental Health, USA; 2016 [cited 2016 Dec 17]. Available from: http://imagej.nih.gov/ij
19. Perez-Filgueira DM, Brayfield BP, Phiri S, Borca MV, Wood C, Morris TJ. Preserved antigenicity of HIV-1 p24 produced and purified in high yields from plants inoculated with a tobacco mosaic virus (TMV)-derived vector. J Virol Methods. 2004;121(2):201-8.
20. Benitez J, Ferrer A, Domínguez Horta MC, Villareal A, Rivera JM, Rodriguez D, et al. One-step purification of genetically engineered HIV-p24 using IMAC. 2001; Int J Bio-Chromatogr 6(3):185-94.
21. Zhenbo X, Zhongyu G, Xihong Z, Youhong Z, Xiaowei H, Wenmei L, et al. Expression and purification of gp41-gp36 fusion protein and application in serological screening assay of HIV-1 and HIV-2. Afr J Microbiol Res. 2012,6(33):6295-9.
22. Narciandi RE, Caballero A, Roque D, Ponce M, Leal V, Novoa LI. Bacterial expression, isolation and purification of a diagnostic recombinant protein representative of HIV-1 gp120 (TAB1). Biotecnol Appl. 1995;12(2):90-1.
23. NEB. Protein data. Bacterial Cells: E. coli or Salmonella typhimurium. Ipswich: New England Biolabs. 2016 [cited 2016 Dec 17]. Available from: https://www.neb.com/tools-and-resources/usage-guidelines/protein-data
24. Marston FA. The purification of eukaryotic polypeptides synthesized in Escherichia coli. Biochem J. 1986;240(1):1-12.
Received in November, 2016.
Accepted in February, 2017.
Duniesky Martínez. Laboratorio de Biología Molecular, Centro de Ingeniería Genética y Biotecnología de Sancti Spíritus. Apartado 83, Sancti Spíritus, CP 60200, Cuba. E-mail: duniesky.martinez@cigb.edu.cu.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}