










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBiotecnología Aplicada
versión On-line ISSN 1027-2852
Biotecnol Apl vol.35 no.1 La Habana ene.-mar. 2018
TECHNIQUE
Advantages of an immunomodulatory assay for the determination of the in vitro potency of human gamma interferon
Ventajas de un ensayo inmunomodulador para la determinación de la potencia in vitro del interferón gamma humano
Luisa G Silega Coma1, Haydee Gerónimo Pérez1, Dayana C Brito López1, Yusnielis Bustamante Pérez1, Iliana Delgado Martínez2, Iraldo Bello Rivero3, Gustavo Furrazola Gómez2, Oscar Cruz Gutiérrez2, Gerardo García Illera1, Mabel Izquierdo López1, Lourdes B Costa Anguiano1
1 Dirección de Control de la Calidad, Centro de Ingeniería Genética y Biotecnología, CIGB. Ave. 31 e/ 158 y 190, Playa, La Habana 11600, Cuba.
2 Dirección de Producción, CIGB, La Habana, Cuba.
3 Dirección de Investigaciones Clínicas, CIGB, La Habana, Cuba.
ABSTRACT
Products containing human interferon human recombinant gamma interferon (IFNγ-hr) as active pharmaceutical ingredients are promising alternatives for treating juvenile rheumatoid arthritis (as Heberon® Gamma R) or skin cancer (e.g. HeberFERON®). Significantly, its development relies on the availability of analytical methods to evaluate the biological activity of the formulation. Therefore, an in vitro immunomodulatory assay was established for quantifying the rhIFNγ biological activity in the referred products, so as to fulfill the European Pharmacopoeia requirements. The average biological potency obtained for several batches using this assay was compared and against an antiviral assay in use. The antiviral assay is based on the rhIFNγ capacity of protecting Hep-2C cells against the viral attack. The immunomodulatory assay assesses the overexpression of Human Leukocyte Antigen-DR (HLA-DR) in COLO-205 cells when they are in contact to the IFNγ-hr. The absorbance readings were processed using a statistical parallel lines program. The immunomodulatory assay was specific for the rhIFNγ effect, with dose-response curves in a sensibility range 800-6.25 IU/mL, equivalent to the range 40-0.31 ng/mL. It was demonstrated that the recombinant human alpha-2b interferon (rhIFNα2b), present in the HeberFERON® formulation, does not interfere in the quantitative determination of rhIFNγ biological activity. Even though both assays detected similar values of biological activity for the different lots tested, the immunomodulatory assay results confirmed its advantages as compared to the antiviral assay.
Keywords: recombinant human gamma interferon, immunomodulatory assay, antiviral assay, Heberon® Gamma R, HeberFERON®
RESUMEN
Los productos biotecnológicos que contienen el interferón gamma humano recombinante (IFNγ-hr) como ingrediente farmacéutico activo constituyen soluciones terapéuticas prometedoras para el tratamiento de artritis reumatoide juvenil (e.g., Heberon® Gamma R) o el cáncer de piel (HeberFERON®). Para su desarrollo es necesaria la disponibilidad de ensayos que permitan medir la actividad biológica de las formulaciones del producto. Por tales razones, se estableció un ensayo inmunomodulador in vitro para cuantificar la actividad biológica del IFNγ-hr en los productos referidos; a fin de cumplir con los requerimientos de la Farmacopea Europea. Además, se compararon los resultados de potencia biológica promedio obtenida para varios lotes usando el ensayo inmunomodulador y el ensayo antiviral actual. El ensayo antiviral, se basa en la capacidad del IFNγ-hr de proteger las células Hep-2C, ante el ataque viral; mientras que el ensayo inmunomodulador evalúa la sobre expresión del Antígeno Leucocitario Humano DR (HLA-DR), en células COLO-205, cuando entran en contacto con el IFNγ-hr. Las lecturas de absorbancia fueron procesadas en un programa estadístico de líneas paralelas. El ensayo inmunomodulador fue específico ante la acción del IFNγ-hr, obteniéndose curvas dosis-respuestas en el rango de sensibilidad entre 800 a 6.25UI/mL, equivalente al rango de 40 a 0.31ng/mL. Se demostró que el interferón alfa-2b humano recombinante (IFNα2b-hr) presente en la formulación del HeberFERON®, no interfiere en la determinación cuantitativa de la actividad biológica del IFNγ-hr. Aunque ambos ensayos detectaron valores similares de actividad biológica, en el total de lotes analizados; los resultados del ensayo inmunomodulador confirmaron sus ventajas en comparación al ensayo antiviral.
Palabras clave: Interferón gamma humano recombinante, ensayo inmunomodulador, ensayo antiviral, Heberon® Gamma R, HeberFERON®.
INTRODUCTION
Third generation recombinant products containing recombinant human gamma interferon (rhIFNγ) as active pharmaceutical ingredients are promising alternatives for treating juvenile rheumatoid arthritis (as Heberon® Gamma R)[1] or skin cancer (e.g. HeberFERON®)[2], chemo-resistant pulmonary tuberculosis, idiopathic pulmonary fibrosis and baso-cellular skin carcinoma, among others [3, 4]. Significantly, to enhance the current biotechnology industry by reaching more competitive standards, the assays must comply with the demands of the regulatory authorities. Determination assays for biological activity of biopharmaceutical products is mandatory, since it directly demonstrates the usefulness of the drug throughout the development and production of the product, as well as in the stability studies [5].
The biological activity of pharmaceutical products is measured by many methods based on Biological Assays or Bio-assays, defined as the analysis of a product for the quantification of biological activity of one or more of its components; it determines its capacity to produce the expected biological potency on living cells (in vitro), expressed in units in relation to the Reference Material (RM) [6].
Two of the most common in vitro biological assays used to determine the biological activity of rhIFNγ are: the antiviral assay (AV) and the immunomodulatory assay. Both assays involve the pre-incubation of cells that are susceptible to cytokines that can induce a specific cellular function [7]. For this antiviral assay, also known as the assay for the inhibition of the cytopathic effect, a cytokine-sensitive cell line is needed; after getting into contact with the sample, a virus is added, which produces a cytopathic effect. The ability of interferon to protect cells from the viral attack is expressed as an increase of the surviving cells as the Interferon dosage increases [8].
There are different combinations of systems (virus-cells) [8] that are available for antiviral assays, although those most appropriate ones should demonstrate the sensitivity of the assay. At the end of the assay, the number of surviving cells is determined with a conventional developing method and the calculation of biological activity through a parallel lines (PL) statistical program. The assay is currently applied at the Biological Assays II Laboratory for Quality Control at the CIGB, using the Hep-2C cell line and the viral species of encephalomycarditis virus.
The immunomodulatory assay measures the ability of rhIFNγ to increase the expression of the Human-Leukocyte-Antigen-DR (HLA-DR), in cell line COLO-205. The quantification of this expression is performed with the use of an ELISA on cells. This immuno-enzymatic assay requires two antigens: one is the primary (monoclonal) antigen that detects the antigens at the cell surface, and the other (polyclonal) antigen, conjugated with the peroxidase enzyme, can detect the primary antigen and transform a specific indicator substrate of the antigenic over-expression.
Data processing is made through the parallel lines method. This is a mandatory assay as ruled by the European Pharmacopoeia for Gamma-1b Interferon [9].
Hence, the Cuban biotechnological industry requires the continuous implementation of quality control techniques for measuring the biological activity of products. Such techniques have to be time feasible and comply with current regulatory requirements. This is very relevant to cope with growing production needs for new pharmaceuticals and to compete with similar products in the world market. Therefore, in this work, an in vitro immunomodulatory assay was established, to determine the biological activity of rhIFNγ in the formulations of Heberon® Gamma R[1] and HeberFERON®.
Establishing and comparing the new immunomodulatory assay (recommended by the European Pharmacopoeia) to the one currently in use, in order to quantify the biological potency of the rhIFNγ, will grant our products a higher quality standard for an efficient response to commercial actions and its domestic and international registration. At the same time, the assay will be of a high methodological, economic and social importance because of its participation in the successful outcomes of this industry.
MATERIALS AND METHODS
Cell lines
Immunomodulatory assay
The COLO-205 line of the European Collection of Authentic Cell Cultures (human colon adenocarcinoma, ECACC 87061208) was selected as a model for the determination of the biological activity of the rhIFNγ, through an immunomodulatory assay (ELISA on cells), based on its ability to express the DR antigen of the MHC class II [10].
Cells were maintained at a range of 3-9 × 105 cells/mL, in the RPMI 1640 (Gibco) medium containing 10 % Bovine Fetal Serum (BFSI) (Capricorn, Australia), 2 mM glutamine and 50 µg/mL gentamicin (Biologicals Industries), incubated for 72 h under culture conditions (37 ºC; 5 % CO2 and 95 % relative humidity). For the assay we used cell growth, both in suspension and anchoring; the passes were made according to the instructions described in the manufacturer’s manual. The assays were carried out between passes 13 and 29 (including the P+5 of the company).
Antiviral assay
Cell line Hep-2C of the European Collection of Authentic Cell Cultures (derived from HeLa, cervix carcinoma, ECACC 85020207) was adapted and maintained at between 1-3 × 104 cells/cm2 in the culture medium MEM CANE (Gibco), containing 10 % BFSI, 2.2 g/L NaHCO3 and 50 µg/mL gentamicin (Biologicals Industries) and incubated for 72 h under the culture conditions mentioned above. The assays were carried out between passes 12 and 23 (including the P+4 of the company).
Virus
Mengovirus (Order Picornavirus; Family Picornaviridae; Genus Cardiovirus; Species: Encephalomyocarditis virus (murine virus) Serotype: Mengo virus (EMCV strain) [11]), was obtained from the American Type Culture Collection (ATCCTM VR-1598). This viral family covers non-enveloped small viral particles, with an icosahedron capsid of 27 to 30 nm in diameter and a single chain RNA genome, with a positive polarity of approximately 7.8 kb, enabling direct translation (functionally equivalent to a messenger RNA), and coding for a simple and single poly-protein of approximately 220 kDa [12]. Master Cell Banks (MCB) and Working Cell Banks (WCB) were made and the infective titer was determined, as well as the multiplicity of infection, for the line of work according to Ramakrishnan [13].
Interferons
The rhIFNγ working reference material (WRM) with code IFNgT-04-0511 (156 000 IU/mL), was prepared in the Stability and Reference Material Laboratory of the Quality Control Division, and further calibrated against the International Reference Material Gxg01-902-535 of NIH. The batches assessed were: 05IFAG703 (the Active Pharmaceutical Ingredient of rhIFNγ with 1.27 mg/mL) and 7001G1 (Heberon® Finished Product Gamma R of 0.5 × 106 IU/mL de rhIFNγ), both produced in 2017, at CIGB. The batches of the stability study also tested were: 05IFAG704-3mY (the Active Pharmaceutical Ingredient of rhIFNγ with 1.25 mg/mL stored for 3 months at –70 °C); batch 4001G1-36mA (Heberon® Finished Product, Gamma R with 0.5 × 106 IU/mL of rhIFNγ) under shelf-life conditions: 5 ± 3 °C for 3 years, and batch 506LL01/2-24mA (Finished Product of HeberFERON®: containing 3.0 × 106 IU/mL of rhIFNα2b + 0.5 × 106 IU/mL of rhIFNγ, under shelf-life conditions: 5 ± 3 °C for 2 years. Batch 4302R1/0 of Heberon® Alfa R was used as the negative control of the specificity assays. The values of biological activity in International Units per milliliter (IU/mL) were expressed in reference to the internal reference material (IRM).
Immunomodulatory assay
The assay was carried out in 96-well microplates according to the protocol described [14] with certain modifications. In this assay, the dose-response curves started at 800 IU/mL or 40 ng/mL of protein. A (primary) monoclonal antibody with a dilution of 1/50 was used, this was the anti-HLA-DR (no.M0746; DAKO, USA) that detected the antigens present on the cell surface. Also a (secondary) polyclonal antibody with a dilution of 1/1500 was employed, which was that of mouse anti-IgG obtained at the Immuno-assay Center through the immunization schedule in sheep and conjugated with peroxidase at the CIGB of Sancti Spíritus, Cuba (CF0351503); this could detect the primary antibody and transform a specific substrate, indicating an antigenic over-expression. The antibodies were first titrated.
The increase in the expression of these antigens was produced separately with two preparations of rhIFNγ: one having a known potency (Reference Material) and another with an unknown potency (the samples). The readings of optic density of the plates at 492 nm were processed by a parallel lines program recommended for the calculation of biological potencies [15]. Assay controls were established, which were: minimum expression control: minimum value of absorbance (cells incubated with the titration medium) and the maximum expression control: the maximum value of absorbance (cells incubated with 1600 IU/mL of the WRM).
Antiviral assay
This assay is based on the ability of Interferons to prevent infection and destruction of Hep-2C cells infected with the Mengo virus through the inhibition of the cytopathic effect.
The assay started after growing the Hep-2C cells with 100 µL per well at a concentration of 1.5 × 105 cells/mL in 96-well plates. After 24 h under growing conditions they were diluted to ensure an initial concentration of 78 to 80 IU/mL or 10 ng, for a final volume of 100 µL (serial dilutions of 1:2 in the MEM CANE 1× medium supplemented with 2 % BFSI and 50 µg/mL gentamicin: AV titration medium), at 37 ± 2 ºC, for 24 hours. The virus with an infective dose in tissue culture at 50 % (TCID50 of 108.4/mL), was added at a rate of 3 viral particles per cell present in the well (Multiplicity of infection (MOI): 3), and it was incubated under the same conditions until the cytopathic effect was observed (90 % cell lysis after 18 to 20 h). Finally, it was developed and the cells were fixed using the colorimetric death exclusion method: violet crystal, which only stains the surviving cells when there is a viral attack. The staining agent was homogenized with 50 µL/well of the 10 % acetic acid and the optic density readings of the plate at 578 nm, were processed by a parallel lines program recommended for the calculation of biological potencies [15].
Controls were established for the assay, i.e. virus control: the minimum values of absorbance (cells incubated with the virus), and the cell control: the maximum absorbance value (cells incubated with the titration medium).
To quantify the rhIFNγ in the HeberFERON® batch, it was diluted at 1/640 in the titration medium and neutralized in the final dilution with 1/50 of the human polyclonal antibody anti-rhIFNα2b (batch from Sancti Spíritus, Cuba n.161 initially titrated) and incubated for 1 h at 37 ºC. To have the dose-response curve of the sample start under the same conditions of the WRM, an additional dilution of 1/10 was carried out, which had all curves start at the range of 78 to 80 IU/mL.
Number of assays and validity criteria for the immunomodulatory and antiviral assays
Biological activity for each one of the samples was measured by two analysts through 4 to 6 assays. At least one plate was used each day per analyst, where the sample was analyzed with two replicates. We therefore obtained 4 to 6 potency values, with which we calculated the geometric mean that corresponded to the final potency value in IU/mL. Each plate used different aliquots/vials of the batch.
The average of the Geometric Variation Coefficient (GVC) was calculated using all the assays of each batch performed by the two analysts, and each one of the validity criteria was analyzed for each assay.
Criteria used to assess the validity of the assays were:
1. Compliance of the parallelism between the dose-response curve of the sample and the reference material, which should be greater than 0.05.
2. Compliance of the linearity of each dose-response curve of the samples and the reference material, which should be greater than 0.05.
3. Compliance of the regression of the slopes of the two curves and the correlation between the doses used for the sample and the reference material, which should equal 0.01.
4. The GVC for the immunomodulatory assay should be equal or less than 20 % [9], while for the AV assay it should be equal or less than 25 %.
5. Compliance with the confidence limits established for each assay, for a probability of 95 %, between 70 and 140 %, for the immunomodulatory assay [9], while for the AV assay it ranges from 64 to 156 %.
The assays that did not comply with any one of these criteria were discarded and they were not included in the final value reported.
Comparison of the biological potencies (average) obtained by both methods
Once the 5 batches (two APIs and three final product batches) were evaluated, using both bioassays in vitro, the average potencies were compared in order to demonstrate the similarity of the biological potencies obtained by both biological methods.
After obtaining the potencies from the individual assays, these were averaged for each batch in the analysis. The following quotients were calculated:
- Average potency differences (ΔP):

- Differences in the extent of the confidence interval of the average potencies (ΔCI):
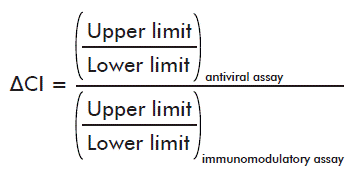
The acceptance criteria took into account that:
- The difference between the average potencies should be at the range of: 0.90 ≤ ΔP ≤ 1.11, for 10 %.
- The difference in the extent of the confidence interval of the average potencies should be of: ΔCI ≥ 0.85.
To demonstrate the similarity of the biological potencies obtained by both biological methods, we considered that the above criteria must be fulfilled in 100 % of the cases.
RESULTS
Specificity of the immunomodulatory assay produced by the rhIFNγ
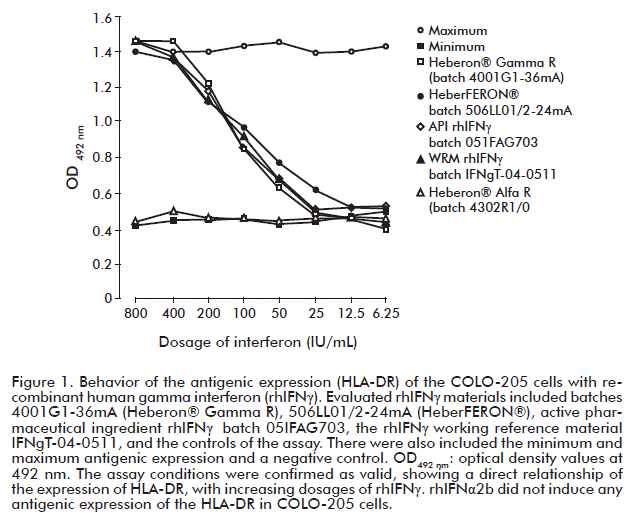
As shown in figure 1, the behavior of response curves to the expression of the HLA-DR antigen by COLO-205 cells incubated for 48 h in the presence of different doses of rhIFNγ (immunomodulatory assay) indicated a very similar behavior between the batches: Heberon® Gamma R (4001G1-36mA), HeberFERON® (506LL01/2-24mA), API (05IFAG703) and WRM (IFNgT-04-0511), which contain rhIFNγ as active principle. These dose-response curves confirm the direct dependence on the over-expression of the HLA-DR antigen as a function of the dose of rhIFNγ administered to cells. It is observed that OD value decreased on diluting the concentration of rhIFNγ, such decrease being directly proportional to the number of antigens expressed, thus confirming the results obtained by Gibson and Kramer [14]. Batches 05IFAG704-3mY and 7001G1, showed similar dose-response curves (data not shown).
The maximum and minimum expression values of these antigens confirm that the procedure for our assay conditions were correct, and that the behavior of the OD values observed in all wells where the dilutions of rhIFNα2b (4302R1/0 of Heberon® Alfa R) were found, were in agreement with the minimum expression of the assay (the dose-response curve was not obtained). This demonstrates that the rhIFNα2b, in the formulation does not interfere in the biological assessment of the rhIFNγ activity.
Validation and precision criteria of the immunomodulatory and antiviral assays
Each one of the validity criteria established for the assays were analyzed. The GVC within assay (between replicates of the same sample) and the average GVC (between assays) of all assays for each batch, carried out by two analysts, were calculated as precision parameters.
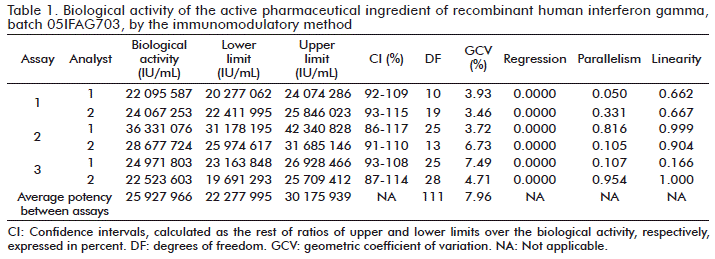
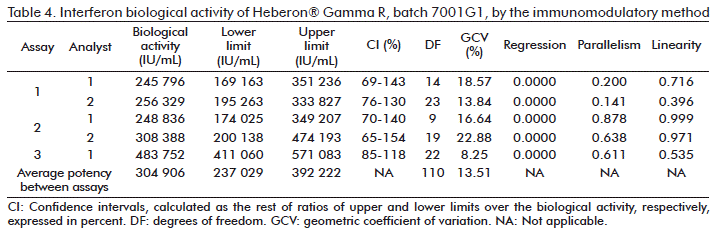
Batches 05IFAG703 and 7001G1 were evaluated in a parallel by the immunomodulatory and antiviral methods. The results for batch (7001G1) of biological activity of independent assays, the average potency, validity criteria and GVC for the analysis of the precision of both methods, are summarized in Table 1, Table 2, Table 3 and Table 4, respectively.
For each independent assay made between analysts to batches 05IFAG703 and 7001G1, by the immunomodulatory and antiviral methods, the validity criteria were fulfilled. The parameters for parallelism and linearity of the dose-response curves of the sample and the WRM were larger than 0.05 in all assays, while the regression of the slopes and correlation between dosages used in the sample and the WRM, were in all cases less than 0.01.
According to the limits of the confidence interval established for the immunomodulatory method, it was demonstrated that the 6 independent assays made with batches 05IFAG703 and 7001G1, had confidence intervals that complied with the established criteria (70 and 140 %, respectively).
The precision values expressed in terms of GVC, according to the specification and variability of the method between replicates in the same plate, showed that the average variabilities of all assays made by both analysts in different days, were less than 20 % (Table 1 and Table 3).
For a total of 5 independent assays performed by the antiviral method to batches 05IFAG703 and 7001G1, the confidence limits for a probability of 95 % are located in the range 64-156 %. The GVC within and between trials for both batches were less than 25 % (Table 2 and Table 4).
Although in all independent assays the confidence intervals and the GVC complied with the criteria established according to the variability of each method (for a probability of 95 %), the widest confidence limits were obtained and a greater variability of the GVC is achieved (values above 10 %) for the AV method for both batches (7001G1; API 05IFAG703), confirming what was expressed in the monograph [9].
Comparison of biological potencies (average) obtained by both methods using newly produced samples of rhIFNγ, and in stability studies
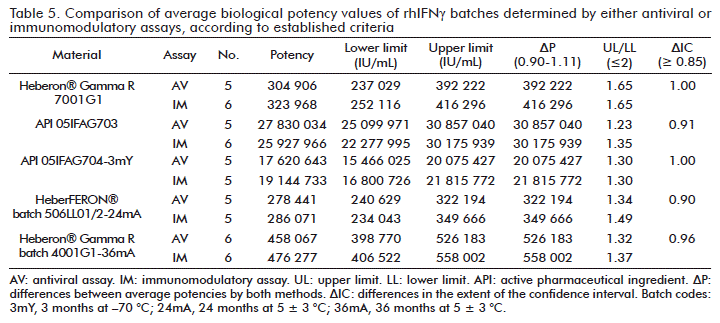
The differences between the average potencies for both methods for the five batches under study are found to be within the appropriate limits for 100 % of the batches analyzed: 0.90 ≤ ΔP ≤ 1.11 (Table 5).
The differences in the extent of the confidence interval of the average potencies between both methods (ΔIC ≥ 0.85), were appropriate in 100 % of the batches in the study (Table 5).
The criteria established for the comparison are fulfilled, showing that both methods detect similar level of biological activity in all batches analyzed.
DISCUSSION
The biological activity of the rhIFNγ can be quantified using a variety of in vitro biological assays, or those based on cells that measure responses, such as antiviral activity [17], anti-proliferative activity [18], or immunomodulating activity [14]. The latter is known as the most potent biological activity, which is found in this cytokine [17].
Although there is a wide variety of cell-virus combinations to evaluate antiviral activity [8, 19], the inhibition of the viral replication of EMCV in cell line Hep-2C is the current method used to determine biological potency and the release of the interferons by the Quality Control Division at CIGB. This assay is cumbersome, it requires long culture periods in a sterile environment, and the need to ensure the conditions and facilities for the management and production of active virus cultures [20].
The inherent variability in infections and viral replication, lead to the decrease of the reproducibility of the antiviral assay [20]; expressed as larger confidence intervals for the antiviral method than for the immunomodulatory method. In view of the need to improve factors such as sensitivity, reliability and the speed of the assay, the immunomodulatory method is recommended for the determination of biological potency, within the rhIFNγ documentation [9].
The immunomodulatory assay makes it possible to determine the biological potency of human recombinant gamma interferon found in different preparations and productive batches produced at CIGB. This assay detects the over-expression of the HLA-DR antigen in COLO-205 cells, which do not structurally express this antigen [17]. Meager, 2002 [14], reports that both type I (rhIFNα2b) and II (rhIFNγ) IFNs, stimulate the MHC class I antigen expression, although only type II IFN can particularly stimulate the de novo synthesis of the MHC class II antigen (HLA-DR).
This aspect gives the trial a high specificity, ensuring that the measured signal only corresponds to rhIFNγ. The assay quantifies the influence of the rhIFNγ in a direct manner on the expression of the HLA-DR and is able to discriminate components such as rhIFNα2b, which is present as part of the HeberFERON® formulation. The behavior of the dose-response curves (Figure 1), confirms these results [14, 19].
Parallelism is a basic requirement [21] to validate the results of the biological activity obtained in the relative potency assays, which can be reported. This parameter is reached in each independent assay for the immunomodulatory and antiviral methods, where in all cases it is larger or equal to 0.05 (Table 1, Table 2, Table 3 and Table 4). The fact that this parameter is reached, demonstrates that both the sample and the standard have the same analyte, and that the WRM and the sample are structurally similar [22]. Therefore, the dose-response curves share the common parameter and are only different in the horizontal displacement [23].
The horizontal displacement between dose-response curves (parallel) of the sample in relation to the standard, implies that the relationship for each dose is constant between curves, which assumes the existence of linearity [24]. This parameter is another one of the validity premises of the statistical calculation of biological potencies. For each independent trial calculated by the immunomodulatory and antiviral methods, the linearity parameter was equal to, or higher than 0.05 (Table 1, Table 2, Table 3 and Table 4).
In both methods, the behavior of the cell response as a function of the concentration is not a straight line, and the data obtained are adjusted using linear regression or the transformation of values [25]. The regression of the slopes of the sample, and the WRM and the correlation between the doses used for the sample and the RM, as a criterion of high significance, was less than 0.01 in all cases, thus confirming the dependence of the cell responses in relation to the concentration of rhIFNγ that is added [23].
The precision is related to the position of the measurements around a mean or central value and it corresponds to the degree of agreement between individual assays when the method is applied repeatedly to multiple aliquots of a homogeneous sample [25, 26]. The within assay precision (between replicates of each dilution of the sample used by the same analyst and on the same day), and the precision between assays (between values of biological activity of each batch calculated by different analysts), expressed in terms of GVC, complied in all cases with the limits established according to the method used (Table 1, Table 2, Table 3 and Table 4) [9, 16].
Any estimation of potency obtained from a biological assay is submitted to a random error because of the intrinsic variability of the biological response. The confidence interval for the potencies is an indication of the precision with which the potency has been estimated in the assay. In other words, it defines the probability range that it contains the true value of the parameter. Generally, in biological assays the calculated confidence limits are of 95 %, so that the expectation is justified that there is a probability (confidence) of 95 % that these limits would include the true potency [25]. The acceptability of this precision depends on the requirements established in the monograph of the preparation under evaluation [27]. For both assays, the intervals calculated complied, in all cases, with the limits established according to the method used (Table 1, Table 2, Table 3 and Table 4).
In the biological activity assays, the potency values calculated for the homogeneous samples of the same product may be variable. For this reason a range is established, within which these values should be found [28]. Studies made by Estévez Carrizo [29], report that for a large variety of drugs, the probability of the error of the method that determines the bioequivalence in relation to a reference, must be controlled. Considering that this risk cannot surpass 5 %, these studies express that, in order to evaluate the bioequivalence of two products, the interval analyzed must be of 80 to 125 %, for the quotient of the average of the products, as the standard criterion (since there is a logarithmic transformation of the data). This corresponds to a range of 20 % for the relative difference between the averages of the products. We established as our comparison criterion a range of relative difference of 10 %: 0.90 ≤ ΔP ≤ 1.11. Within the calculated parameters for the bioequivalence analysis between average potency values, calculated by both methods (immunomodulatory and antiviral), we determined that for both hypotheses the criteria for the batches analyzed were met (Table 5).
The potency of a biotherapeutic product, as a measure of its relative activity with a standard, is a regulatory requirement that enables the confirmation of the correct clinical dose, while it assesses stability, productive consistency or/and detects changes during the manufacturing process [20]. In this study, the biological potency determined by the immunomodulatory and antiviral methods for each batch was similar. It was demonstrated that the immunomodulatory trial is applicable to evaluate biological potency in fresh samples and under shelf-life conditions: 5 ± 3 °C, thus ensuring its clinical use during this period. Gibson and Kramer [14] also demonstrated that this approach could contribute to quality control. As a study case they evaluated the bioactivity of both rhIFNγ preparations stored for two years at 25 and 45 °C, they quantified antiviral activity by the inhibition of the cytopathic effect provoked by the EMCV method in cell line A549 (human lung carcinoma) and the immunomodulatory activity, quantifying the expression of the HLA-DR in the COLO-205 line. Both methods demonstrated the loss of activity in the preparations in relation to the control stored at -70 °C, detecting similar biological potencies for each preparation using the antiviral and immunomodulatory methods, which confirm our results.
It is, however, necessary to include batches under accelerated conditions in future studies, thereby ensuring the use of the drug when there are unexpected failures in the cold chain, and always considering that the method must be able to detect abnormal potencies due to changes in the molecule.
This immunomodulatory method is less variable, and more precise and specific for rhIFNγ, thereby confirming the results obtained. Furthermore, to carry out the trial it requires a smaller number of biological agents (it does not include the risk of handling a viral agent), and less time is taken for the assay compared to the AV method.
Both assays require the use of basic or routine laboratory equipment. As modifications of the original method [14], the immunomodulatory assay does not include automatic plate washers, this is done manually, and primary and secondary antibodies were modified, but good results were attained.
In short, this paper confirms the advantages of the immunomodulatory assay compared to the AV assay. It was shown to be applicable in the quantification of the biological potency of the Cuban gamma interferon, obtaining values that are very similar to the antiviral method now in use for the release, by the Quality Control Division, of products such as Heberon® Gamma R and HeberFERON®. Its use is recommended as a routine assay after concluding the validation, according to national and international regulations, with the aim of ensuring reproducible and reliable results, according to its purpose.
Methodologically, the achievement of our goals is relevant for other laboratories and institutions that can apply the method, thereby enriching the professional experience of analysts when facing the challenges of new technologies within Cuban biotechnology. This analytical technique can form part of technology transfer packages, granting them notable economic importance. It should be pointed out that any contribution enabling the availability of novel drugs, with proven quality and safety, will have a positive impact on the health of the population and will therefore be of considerable social importance.
CONFLICTS OF INTEREST STATEMENT
The authors declare that there are no conflicts of interest.
REFERENCES
1. CIGB. Registro Sanitario, Heberon® Gamma R, 2007. (Interferón gamma hu-rec). No. B-07-199-L03. La Habana: CIGB; 2007.
2. CIGB. Registro Sanitario, HeberFERON®, 2016. (Interferón alfa 2b hu-rec + Interferón gamma hu-rec). No. B-16-156-L03. La Habana: CIGB; 2016.
3. Cayón I, González L, García I, Rosas C, Gassiot C, García E, et al. Gamma Interferon and prednisone decreasing-dose therapy in patients with Idiopathic Pulmonary Fibrosis. Biotecnol Apl. 2010;27(1):29-35.
4. Rojas Rondón I, Duncan Roberts Y, Gómez Cabrera CG, Ramírez García LK, Vigoa Aranguren L, Hernández Rodríguez R, et al. Administración del HeberFERON en el carcinoma basocelular palpebral a propósito de 2 casos. Bionatura. 2016;1(2):71-4.
5. United States Pharmacopeial Convention. United States Pharmacopeia. Vol 1: <1032> Design and development of biological assays. United States Pharmacopeial Convention 39, National Formulary 34, (USP 39- NF 34). Rockville, Maryland, USA: United States Pharmacopeial Convention; 2016.
6. United States Pharmacopeial Convention. United States Pharmacopeia. Vol 1: <1030> Biological Assays Chapters- Overview and Glossary. United States Pharmacopeial Convention 39, National Formulary 34, (USP 39- NF 34), Rockville, Maryland, USA: United States Pharmacopeial Convention; 2016.
7. Whiteside TL. Cytokine Assays. BioTechniques. 2002;33:4-15.
8. Council of Europe and European Pharmacopoeial Commission. European Pharmacopoeia (EP). Vol 1. <5.6> Assay of Interferons. Prepared under Council of Europe and European Pharmacopoeial Commission 9th ed. Strasbourg, France: EEPC; 2016.
9. Council of Europe and European Pharmacopoeial Commission. European Pharmacopoeia (EP). Monography on Interferon Gamma 1b Concentrated. Prepared under Council of Europe and European Pharmacopoeial Commission 9th ed. Strasbourg, France: EEPC; 2016.
10. Matsushita K, Takenouchi T, Shimada H, Tomonaga T, Hayashi H, Shioya A, et al. Strong HLA-DR antigen expression on cancer cells relates to better prognosis of colorectal cancer patients: Possible involvement of c-myc suppression by interferon-γ in situ. Cancer Sci. 2006;97(1):57-63.
11. Racaniello, VR. Picornaviridae: The viruses and their replication. In: Knipe DM, Howlez PM (Editors). Fields' Virology. 6th edition. Volume 1. Chapter 16. Philadelphia: Lippincott Raven Publishers; 2013. p. 453-89.
12. Carocci M, Bakkali-Kassimi L. The Encephalomyocarditis virus. Virulence. 2012;3(4):351-67.
13. Ramakrishnan MA. Determination of 50% endpoint titer using a simple formula. World J Virol. 2016;5(2):85-6.
14. Gibson UE, Kramer SM. Enzyme-linked bio-immunoassay for IFN-gamma by HLA-DR induction. J Immunol Methods. 1989;125(1-2):105-13.
15. Council of Europe and European Pharmacopoeial Commission. European Pharmacopoeia (EP). Vol 1. <5.3> Statistical Analysis of Results of Biological Assay. Prepared under Council of Europe and European Pharmacopoeial Commission 9th ed. Strasbourg, France: EEPC; 2016.
16. Malek Sabet N, Masoumian RM, Nasiri-Khalili MA, Maghsoudi N, Sami H, Saeedinia A, et al. The structural characterization of recombinant human interferon gamma. J Biol Sci. 2008;8(6):1087-91.
17. Bello-Rivero I, Torrez-Ruiz Y, Blanco-Garcés E, Pentón Rol G, Fernández-Batista O, González LJ, et al. Construction, purification and characterization of a chimeric TH1 antagonist. BMC Biotechnol. 2006;6:25:1-13.
18. Meager A. Biological Assay for Interferons. J Immunol Methods. 2002;261(1-2):21-36.
19. Moore M, Ferguson J, Burns C. Applications of cell-based bioassays measuring the induced expression of endogenous genes. Bioanalysis. 2014;6(11):1563-74.
20. Callahan JD, Sajjadi NC. Testing the Null Hypothesis for a Specified Difference: The right way to test for parallelism. BioProcessing J. 2003;2(2):71-7.
21. Fleetwood K, Bursa F, Yellowleees A. Parallelism in practice: approaches to parallelism in bioassays. PDA J Pharm Sci Technol. 2015;69(2):248-63.
22. Finney DJ. Statistical Methods in Biological Assays. London: Charles Griffin & Co. Ltd.; 1964.
23. Yu B, Yang H. Evaluation of different estimation methods for accuracy and precision in biological assays validation. PDA J Pharm Sci Technol. 2017;71(4):297-305.
24. United States Pharmacopeial Convention. United States Pharmacopeia. Vol 1: <1225> Validation of compendial procedures. United States Pharmacopeial Convention 39, National Formulary 34, (USP 39-NF 34). Rockville, Maryland, USA: United States Pharmacopeial Convention; 2016.
25. United States Pharmacopeial Convention. United States Pharmacopeia. Vol 1: <111> General Chapter Design and Analysis of Biological Assays. United States Pharmacopeial Convention 39, National Formulary 34, (USP 39-NF 34). Rockville, Maryland, USA: United States Pharmacopeial Convention; 2016.
26. Mire-Sluis A, Page L. Quantitative cell line based bioassays for human cytokines. J Inmunol Methods. 1995;187:191-9.
27. Estévez Carrizo FE. Estudios de bioequivalencia: enfoque metodológico y aplicaciones prácticas en la evaluación de medicamentos genéricos. Rev Med Uruguay. 2000;16:133-43.
Received in October, 2017.
Accepted in March, 2018.
Luisa G Silega Coma. Dirección de Control de la Calidad, Centro de Ingeniería Genética y Biotecnología, CIGB. Ave. 31 e/ 158 y 190, Playa, La Habana 11600, Cuba. E-mail: luisa.glenda@cigb.edu.cu.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}