










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal

Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkBiotecnología Aplicada
versão On-line ISSN 1027-2852
Biotecnol Apl vol.35 no.2 La Habana abr.-jun. 2018
REVIEW
Present and future of CRISPR/Cas systems in Biotechnology
Presente y futuro de los Sistemas CRISPR/Cas en Biotecnología
Inmaculada Viedma
Facultad de Enfermería de la Universidad Católica de Murcia, España.
ABSTRACT
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats), along with the Cas endonuclease genes, form the CRISPR/Cas system. These systems were discovered as a defense mechanism in the Bacteria and Archaea domains, in which DNA from a pathogen, such as a bacteriophage, is incorporated between repeated palindromic sequences and later transcribed into an RNA known as crRNA. Upon subsequent infection s with the same pathogen, the crRNA coupled with Cas targets the transcribed foreign RNA sequences and silences them. The endonucleolytic activity and sequence specificity of the CRISPR/Cas system have been harnessed in genetic engineering to activate or repress genes, to induce point mutations, and to alter sequences through homologous recombination. CRISPR/Cas has also been used to evaluate cellular physiology through the simultaneous activation or repression of various genes. In this article, it is reviewed the history and mechanism of action of the CRISPR-Cas system, its potential applications in cell and gene therapy, and the bioethical implications of the latter.
Keywords: CRISPR/Cas, biotechnology, genetic engineering, gene therapy, molecular biology, genetic expression, bioethics.
RESUMEN
Las secuencias CRISPR (Repeticiones Palindrómicas Cortas y Regularmente Espaciadas y Agrupadas) junto con los genes que codifican para las endonucleasas Cas, forman los llamados sistemas CRISPR/Cas. Los CRISPR se descubrieron como un sistema presente en los Dominios Bacteria y Archaea que les confiere inmunidad frente a patógenos. Cuando un organismo patógeno infecta a una bacteria ocurre una inserción, en las agrupaciones CRISPR del hospedero, de nuevos espaciadores procedentes del genoma del virus y esta integración confiere inmunidad específica frente al invasor. Gracias a su capacidad de reconocimiento de secuencias específicas y a su actividad endonucleasa, el sistema CRISPR/Cas se ha utilizado en ingeniería genética con el fin de activar o reprimir la expresión de determinados genes. Este artículo recoge la situación actual de estas técnicas, las opciones de futuro que ofrecen y la valoración desde el punto de vista de la bioética de las posibles aplicaciones terapéuticas.
Palabras clave: CRISPR-Cas, biotecnología, ingeniería genética, terapia génica, biología molecular, expresión génica, bioética.
INTRODUCTION
“Gene editing” is a recently coined term that refers to a genetic engineering technique in which a DNA fragment is inserted, deleted or replaced in the genome of a target cell by using nucleolytic enzymes, otherwise known as nucleases. These nucleases are used to introduce double-stranded breaks (DSB) at specific locations in the genome, and the resulting DSB are then repaired either by non-homologous end joining (NHEJ) or homology-directed repair (HDR), resulting in controlled modifications (editing).
Two nuclease systems had previously been used for this purpose:
- Zinc-finger nucleases (ZF nucleases), engineered by fusing DNA-binding zinc-finger domains to the catalytic domain of the Fok I restriction endonuclease [1].
- Transcription Activator-Like Effector-based Nucleases (TALENs), based on fusions of the catalytic domain of the Fok I restriction endonuclease to a sequence-specific DNA binding domain [2].
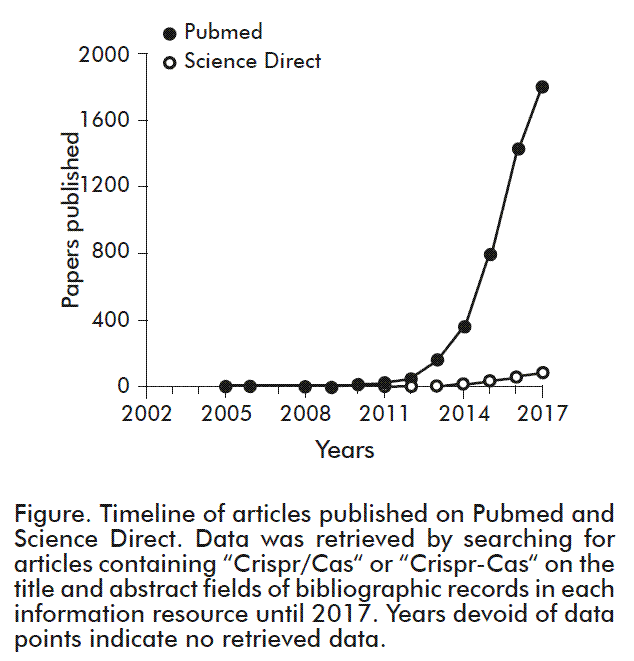
These systems, however, were labor-intensive and expensive. For instance, developing a zinc-finger nuclease required 4-5 years of work at an approximate cost of 30.00 €, while the use of TALENs requires 3-4 months of preparatory work, at a cost of some 10 000 €. Thus, the advent of CRISPR/Cas systems, which only require 2-3 weeks of work at a cost of 20-30 €, represented a giant leap forward for the gene editing community, as illustrated by the rapid growth in the number of publications using this technique since its initial description (Figure). Therefore, this review is aimed to describe the history and mechanism of action of the CRISPR/Cas system, its potential applications in cell and gene therapy, and the bioethical implications of the latter.
DISCOVERY OF CRISPR/Cas
In 1987, Ishino et al. [3] described the existence of clusters of repeated sequences within the genome of Escherichia coli. Further research led to the description of similar clusters in the genomes of Shigella dysenteriae and Samonella enterritidis [4], and this finding was later repeated in many different microorganisms. Tandem 30 to 34 bp long repeats interspersed by non-repetitive 35 to 39 bp sequences were found in the archaeal species Haloferax mediterranei [5,6], and were first denominated Short Regularly Spaced Repeats (SRSR) [7]. SRSR were found to be present in almost half of all bacterial genomes and practically all archaeal genomes sequenced at that moment. In 2002 Jansen and Mojica jointly agree to denote these clusters by the acronym CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) [8].
Although the role of these sequences was unknown at the time, it had been determined that the entire locus was transcribed as a precursor pre-crRNA which upon further processing yielded smaller fragments denoted as crRNA [9].
Concurrently with all this work, Jansen et al. described in 2002 four genes associated with CRISPR clusters, which they denominated cas, for CRISPR-associated [8]. These genes were found to code for nucleases. A thorough biochemical, structural and functional characterization of these proteins followed, performed by a group under the leadership of Jennifer A. Doudna. Their work determined that cas genes were indeed part of the CRISPR system, and assigned putative roles to the gene products of individual cas members [10].
In 2005, Mojica [11] and Pourcel [12] independently discovered that the spacers of the CRISPR clusters were homologous to DNA sequences from bacteriophages, foreign chromosomal fragments and non-transmissible plasmids. They cleverly observed that bacteria bearing these spacers could not be infected by bacteriophages carrying homologous sequences, leading to the deduction that CRISPR/Cas systems probably constituted an adaptive defense system in bacteria and archaea against the introduction of foreign DNA. Bolotin found a conserved sequence adjacent to the protospacer that acted as a leader [13], which was later denominated PAM (Protospacer Adjacent Motif). The PAM sequence is known to play an important role in the operation of several CRISPR/Cas systems [14].
Further advancement in our knowledge of the system was made possible by the team of Emmanuelle Charpentier, which identified small RNAs, denominated tracrRNA, which were found to be necessary for the generation of crRNA in CRISPR/Cas systems [15].
MECHANISM OF ACTION OF CRISPR/Cas SYSTEMS
The nomenclature used to classify CRISPR/Cas systems, which is based on the degree of sequence similarity and architectural organization of CRISPR/Cas loci, groups them into three (I, II and III) types [16] and several subtypes (denoted by a letter) according to the presence of specific proteins. A total of 10 cas genes have been described. Each CRISPR/Cas type contains two universally distributed cas1 and cas2 genes (although their universality is controversial) and a specific cas gene: type I clusters have cas3, type II clusters have cas9, and type III clusters have cas10. The remaining cas genes (4 to 8) are present on at least two types.
As mentioned above, the spacer sequences are homologous to DNA sequences of viral origin and to other genetic elements that may potentially invade the host organism. These spacer sequences are currently known as protospacers [9, 10]. It has been shown that microorganisms bearing protospacers homologous to a sequence present on an infectious agent cannot be further infected by that agent [11], leading to the hypothesis that CRISPR operated as an adaptive defense system of microorganisms against invading DNA elements [11, 12].
CRISPR/Cas systems have so far been found in the Bacteria and Archaea domains. The active acquisition of immunity against a specific bacteriophage was first demonstrated in 2007, in experiments on Streptococcus thermophilus. There was detected the insertion into the CRISPR cluster of the host of new spacers derived from an infecting bacteriophage, which showed that the bacteria carrying the modified CRISPR cluster were resistant to subsequent infections with the same bacteriophage [17].
Although the exact mechanism of action of CRISPR/Cas system changes depending on the specific CRISPR/Cas type or subtype, a common operating model can still be discerned, consisting of three stages: acquisition, expression and interference. During the acquisition stage, the system incorporates spacers derived from an invading genetic element that will later serve to target that element for interference. For this purpose, the cas proteins scan the invading DNA, looking for a short nucleotide motif denominated PAM, and cut out an adjacent fragment. This fragment is usually integrated into the location closest to the leader sequence of the CRISPR cluster of the host.
Next comes the expression stage, when the RNA sequences encoded into the CRISPR clusters are transcribed and the gene products of the cas genes are expressed. The CRISPR locus is transcribed beginning from the leader sequence, forming a pre-crRNA that will be further processed to yield crRNA molecules [18], which will serve as guides during the next stage.
In the third stage, interference, the crRNA is used as a guide to let the Cas proteins bind specifically to the invading DNA and degrade it.
APPLICATIONS OF CRISPR/Cas SYSTEMS
Although practical applications exist of several different CRISPR/Cas systems, the CRISPR/Cas9 pair has received by far the most use owing to several advantages. We will therefore center our explanation on this particular example.
Components and operation mode
The CRISPR/Cas9 system employs two components:
1. The Cas9 enzyme. This nuclease has a bilobular structure where one lobule, denominated REC (for recognition) binds the target and the other, denominated NUC (for nuclease) produces a double-stranded break [19]. The NUC lobule in turn exhibits two structural domains denominated RuvC and HNH based on homologies to structural domains from previously known nucleases, and a PAM recognition domain denoted PI.
2. A duplex RNA formed by a crRNA and a crRNA-transactivating tracrRNA, known under the more general term of guide RNA. The crRNA binds via base pairing to its target on the DNA or protospacer along a homology stretch of 20 nucleotides. The tracrRNA molecule is necessary for the binding of Cas9 to the guide RNA and to maintain the latter in a conformation adequate for the interaction between the crRNA and its target. Its presence, thus, is absolutely required for the operation of this genomic editing technique [20].
The crRNA/tracrRNA hybrid can be formed as a duplex between independent molecules, as found naturally, or as a single-molecule chimaera formed by joining both molecules using a stem and loop. In the latter case it is known as a sgRNA (single guide RNA). This sgRNA exhibits all the essential characteristics of a guide RNA, such as the presence of a 20 nucleotide sequence at its 5´ end that hybridizes to the protospacer in the DNA, and a double-stranded structure in its 3´ end facilitating the binding between the sgRNA and Cas9 [20, 21].
The PAM sequence is not part, structurally speaking, of the CRISPR/Cas tool, but plays an essential role in the process. PAM sequences consist of a series of nucleotides residing at the 3´ region of the target DNA strand that do not bind the sgRNA. The presence of this motif is essential for the recognition by Cas9 of its cleavage site, as Cas9 cleavage is a two-part process in which the endonuclease, together with its associated sgRNA, first recognizes PAM to form a Cas9-DNA complex, and only then the DNA strands are melted and the Cas9 complex starts scanning for its target sequence [22]. PAM recognition takes place, as mentioned above, through the PI domain of Cas9, i.e. is not mediated by sgRNA. The exact composition of PAM sequences varies according to the organism from which the specific Cas9 nuclease is isolated, enabling the independent and simultaneous modification of several sites of the locus of a cell.
Construction of the CRISPR/Cas system and its introduction into the target cell
The oligonucleotide sequences coding for both components of the tool only have to be inserted in DNA form. Both components, Cas9 and the sgRNA, are usually contained within a single expression vector, although they can be present in separate vectors. Most frequently, they are introduced into a single vector [23].
In 2015, Sakuma and Yamamoto demonstrated that it was possible to modify the delivery vector so as to code simultaneously for seven separate sgRNAs, obtaining a CRISPR/Cas construct that, upon introduction into the target cell, expressed a nuclease active simultaneously against seven different targets. In other words, the tool can be modified to code for as many different target specificities as sgRNA molecules are coded by the delivery vector [24].
Once the delivery construct has been obtained, it is necessary to choose an optimal delivery method. The most common options are biochemical, physical or virus-mediated transfection [25-27].
Applications of CRISPR/Cas in genome editing
From 2011 on, awareness of the potential biotechnological applications of the CRISPR/Cas systems began to spread. The three main components of type II CRISPR systems (Cas9, crRNA and tracrRNA) had already been characterized, and a frantic race took place between different research groups that were trying to turn these systems into a genome-editing nuclease whose sequence specificity was dictated by the presence of a guide RNA.
In 2012, Jennifer A Doudna and Emmanuelle Charpentier (who in 2015 were granted the Princess of Asturias award for Technical and Scientific Research to honor their work on developing the CRISPR/Cas system as a gene editing tool) joined forces to dissect, through in vitro experiments, which elements of Type II CRISPR/Cas systems were absolutely required for the sequence-specific cleavage of DNA sequences. They found that only three elements (Cas9, a guide crRNA and a tracrRNA that pairs with the CRISPR segment of the crRNA, or a single chimeric sgRNA) were necessary to generate double-stranded breaks in DNA fragments containing a sequence complementary to the spacer contained within the crRNA. These two researchers were the first to publicly and explicitly acknowledge, in a paper published in Science, the potential application of this system for genome editing. Thus CRISPR/Cas as a gene-editing tool was born [27].
This powerful tool, which will undoubtedly transform radically the medical landscape of the 21th century, can be used for different purposes:
Gene disruption
CRISPR/Cas9 can be used to disrupt genes by generating insertions or deletions. For this purpose, a double-stranded break (DSB) is generated by Cas9 at the target site, which is then spontaneously repaired by the DNA repair system present in almost all cell types [28]. This repair machinery uses either homology-directed repair (HDR) or non-homologous end joining (NHEJ), depending on whether a repairing template is available or not.
If no template is available and the cellular repair system therefore uses NHEJ, random nucleotide additions or eliminations may take place at the ends of the fragments to be joined which may, if the target site happens to reside in a protein-coding sequence, alter the reading frame of this coding sequence. Such an alteration changes the amino acid sequence of the resulting gene product and usually leads to the premature insertion of a stop codon, generating a loss-of-function mutation. These mutations are random and irreversible.
On the other hand, if the objective of the experiment is to repair or modify in a specific way a nucleotide sequence, it is necessary to force the cellular machinery to follow the HDR pathway by providing a repair template containing the desired change. This is usually achieved by co-transfecting a single-stranded oligonucleotide with the template sequence together with the plasmid(s) coding for Cas9 and the sgRNA. In the template, the region containing the nucleotide changes to be introduced at the DSB site must be flanked by stretches homologous to the sequences sur-rounding the site in the endogenous DNA [29].
The efficiency of HDR is not very high, which has prompted a number of research groups to work on the optimization of this technique, either by synchronizing the cell cycle of the culture to be able to perform the transfections at the cell cycle stage at which HDR is most active, or by jamming the components of the NHEJ machinery either via inhibitor compounds or by genetic engineering. Still, it should be noted that even in a best-case scenario the cleavage efficiency of Cas9 is much higher than the efficiency of HDR, and therefore there will always be a significant fraction of repaired DSB that will have undergone NHEJ. Therefore, the presence of the desired modification must always be verified experimentally after using HDR [30].
Inversions and translocations
The ability to generate directed chromosomal translocations, which CRISPR/Cas9 provides, is useful for the study of certain diseases. In this application, two sgRNAs are designed that can be used to generate DSB in two loci sitting in non-homologous chromosomes, followed by NHEJ repair. This process can be performed in vivo.
In the case of inversions the same procedure is followed, but choosing two sgRNAs that simply target two different loci in the same chromosome. The expected result, in this case, is an inversion of the fragment flanked by the target sites [31].
Unfortunately, the repair of two DSB to generate a chromosomal translocation is a rather inefficient process that takes place at frequencies of ~10–3. A procedure has therefore been developed whereas the cells bearing HDR-generated translocations are selected by introducing an antibiotic resistance marker flanked by two LoxP sites, and these in turn are flanked by sequences complementary to the generated fragments that the technique intends to join. Thus, cells in which the translocation has taken place bear the antibiotic resistance marker and can be adequately selected, eliminating the marker later by Cre-mediated site-specific recombination [32].
Regulation of gene expression (activator-repressor)
CRISPR/Cas9 can also be harnessed to either activate or repress gene expression (techniques known respectively as CRISPRa and CRISPRi). These procedures employ a mutated Cas9 with inactive nuclease domains whose target binding specificity remains unaltered (dead Cas9, or dCas9) [33], which is fused to different effector domains. If transcriptional activation is sought, these domains will be transcriptional activator domains. If, on the other hand, the objective is transcriptional repression, there are two options. One is the use of dCas9 by itself, which will produce a knockdown, or if a stronger repressor effect is desired, dCas9 can be fused to transcriptional silencing do-mains, which will result in a transcriptional knockout. In order to ensure the success of the procedure, the sgRNA to be used must target a site as close as possible to the promoter of the gene of interest, to facilitate the operation of any transcriptional activators or silencers fused to dCas9. Unlike the use of CRISPR/ Cas to repair or disrupt genes, this application (transcriptional modulation) does not result in definitive or permanent changes in the transfected cell, as it does not change the sequence of the gene whose regulation is sought.
Sequence-specific chromosome imaging
In this application, dCas9 is fused to a fluorescent protein (EGFP, for instance) and a sgRNA that targets the fusion protein to the desired chromosomal location. The physical location of the targeted loci can then be visualized by fluorescence microscopy [34].
DNA methylation and demethylation
Methylation is one of the mechanisms employed by mammals to control gene expression. A CRISPR/ Cas system has been recently developed that enables the selective in vivo methylation or demethylation of specific sites on the genome of the transfected cell. It employs a fusion of dCas9 with the Tet1 or Dnmt3a enzyme domains to demethylate or methylate, respectively, the target DNA site [35].
Development of cellular models
The generation of mutant clones or knockouts through classical homologous recombination-based techniques has produced through the years a sizable number of very useful isogenic cell lines where the only difference between the parental and mutant lines is a minimal, defined mutation, enabling the study of the function of the disrupted or modified gene. These procedures, however, are highly demanding technically, expensive and labor-intensive, and have always constituted a bottleneck for the generation of isogenic cell lines.
This situation has changed with the advent of CRISPR/Cas technologies, which enable the generation of isogenic human cell lines for comparative genomics purposes in a fast, cheap and relatively simple manner. Likewise, the ability to perform knock-ins of mutant alleles by HDR has enabled the research community to easily test the effects of any identified disease-associated mutations in isogenic backgrounds [36].
Genetic screening
Before CRISPR/Cas was available, loss-of-function (LOF) and gain-of-function (GOF) screenings were performed with RNAi-based libraries of repressed genes and cDNA-based libraries of overexpressed genes. This situation had a number of disadvantages (RNAi generates incomplete knockdowns (false negatives) in a significant proportion of cases and has a relatively high rate of off-target effects, generating false positives). All these have been minimized through the use of CRISPR/Cas. The system currently most used for this purpose employs pooled lentiviral CRISPR libraries, consisting of a heterogeneous population of lentiviral transfer vectors, each containing a single sgRNA directed to a single gene of the organism under study [36].
In cancer research CRISPRa has been used to identify genes that constitute positive or negative regulators of the proliferation of malignant cells, and also to dissect which genes, when overexpressed, may produce a phenotype of resistance to a specific anticancer drug [37].
Some laboratories have used these techniques to model colon cancer [38].
Disease models
Animal models of some human diseases have been developed, such as: pig models of Parkinson’s disease [39], primate models for Duchenne’s muscular dystrophy [40], immunodeficiency in mice and zebra fish [41, 42], and for cardiovascular diseases [43].
Human gene therapy
Despite the latest attempts for application and the most restrictive ones, gene therapy approaches have been reported. For instance, the treatment of a patient with lung cancer [44], as well as the approval in USA of using CRISPR/Cas to treat cancer patients requiring the infusion of T-cells [45].
One of the latest achievements of the application of CRISPR/Cas technology to gene therapy was the excision of an HIV-1 provirus in animal models [46].
The Izpisua’s group has used a CRISPR “scalpel” to edit the MYBPC3 gene in human embryos to avoid mosaicism. When one of the two copies of this gene is mutated, it may cause hypertrophic cardiomyopathy, a disorder that can produce sudden death and heart failure [47].
Limitations, problems and inconveniences remaining unsolved
One of the most concerning limitations of the CRISPR/Cas technology is the relatively high rate, in some cases, of non-point mutations and undesired chromosomal translocations stemming from the induction of DSBs on sites unrelated to the intended target, also known as off-target effects [48]. While this relative lack of specificity may pose an evolutionary advantage within the biological context in which the system evolved (enabling the defense against invading hypervariable plasmid or viral DNA) it hinders considerably the application of the technique for genome modifications, especially in a clinical setting. The number of off-targets varies depending on the number and position of mismatches on the off-target site, and the influence of epigenetic variations among different cell types cannot be discarded as a source of further variation in this respect [49].
This problem is being tackled using several different approaches (Table).
Potential future applications of CRISPR/Cas technologies
The advent of this technique has opened up many different future possibilities within every field of genetic engineering.
However, it is in the field of medicine where this system has raised most expectations, owing to its many potential applications. In disorders with an underlying genetic cause or genetic risk factors, repair mechanisms induced by Cas9 might be harnessed to, ideally, eliminate undesired mutations. For instance, the therapy of disorders caused by the presence of invasive genomes or dominant negative mutations might be approached through CRISPR/Cas-mediated gene disruption. The ability of Cas9 and Cas9-based protein fusions to effect transcriptional activation or repression could also be harnessed to repress the transcription of oncogenes or viral receptors in the host cells or to activate the transcription of tumor-suppressor or globin genes [58].
Another field of knowledge that stands to benefit greatly from the application of CRISPR/Cas technologies is the biotechnological industry. There, CRISPR/Cas can be used to implement or modify new biosynthetic or catabolic routes in production strains so as to increase the yields of relevant processes (manufacturing of biofuels, biomaterials, etc). The technology can also be applied to crop improvement, an application with obvious implications for the food industry- and even to the development and production of drugs and/or cosmetics by living organisms. In practice, CRIPR/Cas technologies are being used in almost all fields of the biotechnological industry.
ETHICAL IMPLICATIONS OF THE APPLICATION OF CRISPR/Cas SYSTEMS
Pre-existing guidelines and moratoria regarding new genetic modification techniques
From its very beginnings, genetic engineering has attracted the attention of bioethicists, who have argued about and discussed its procedures and, especially, its applications. The first reaction of the scientific world once the first genetic engineering procedures were developed (RNA-to-DNA transcription using reverse transcriptase, in vitro recombination using restriction endonucleases, plasmid and phage vectors, etc.) was one of caution and prevention. Even before any risks had been properly assessed, potentially dangerous experiments were temporarily proscribed in July 1974, in what became known as the Berg moratorium. The signees to this moratorium asked for the temporary suspension of specific types of experiments, the organization of an international meeting to discuss the safety issues thought to be associated with the new genetic recombination techniques, and the implementation of a regulatory body under the management of an important scientific organization.
This declaration, which found quick worldwide acceptance, led to a February 1975 meeting in Asilomar under the title of “International Conference on Recombinant DNA” that gathered scientists from all over the world. After considerable discussion, the meeting produced a regulatory project containing the then-new concept of biological containment.
In July 1976 the National Institutes of Health (NIH) issued a very restrictive set of regulations pertaining recombinant DNA that found worldwide resonance. The main objective of these regulations, which stemmed from the Asilomar conference, was to prevent the escape of recombinant organisms from the laboratory.
A different milestone with similarly important bioethical implications regarding the right to privacy and the manipulation of human genes was the sequencing, years later, of the human genome [59]. Member countries of the European Union, aware of the importance of paying respect to the human being as a person and as a member of the human species, and of the need to guarantee human dignity, prepared and signed the Oviedo Convention [60]
(Convention for the Protection of Human Rights and Dignity of the Human Being with regard to the Application of Biology and Medicine). This project in turn led UNESCO to publicly state its position on this matter, resulting in the Universal Declaration on the Human Genome and Human Rights [61] (UNESCO, Paris, November 11, 1997).
Ethical dilemmas created by the CRISPR/Cas system
There is no shortage of ethical dilemmas created by the application of CRISPR/Cas systems, going from their use in plants to be used for human nutrition to the development of transgenic insects to fight malaria [62], with a myriad application in both basic research and medicine in between.
Concerns among the scientific community about the ethical implications of CRISPR/Cas technologies, which had already been growing, peaked with their application, by a research group at the Sun-Yat-Sen University in Guangzhou, China, to edit the HBB gene coding for the β-chain of hemoglobin in pre-implanted human embryos [63]. Actually, this research team used nonviable embryos, with three pronuclei that cannot be implanted. The Chinese scientists found that CRISPR/Cas could be used to edit the HBB gene, but the efficiency of HDR-based repair was low, and the embryos where the genetic modification was successful exhibited numerous malformations. Also, there were errors in the excision process, and the endogenous HDB gene from hemoglobin, which is an HBB homologue, competed with the provided exogenous repair template, producing a number of adverse mutations. The Chinese team concluded: “Taken together, our work highlights the pressing need to further improve the fidelity and specificity of the CRISPR/Cas9 platform, a prerequisite for any clinical applications of CRSIPR/Cas9-mediated editing” [63].
Before the paper detailing their work was published (it was rejected both by Science and Nature), two groups of scientists wrote editorials to Nature [64] and Science [65] expressing their concerns about this research. The group behind the editorial signed by Lanphier requested a moratorium on the edition of the human germ line, based on the high risks and relatively low benefits afforded by hereditary genetic modifications. Genome editing research done in animals has shown that it is possible to activate or inactivate genes on an embryo; a process that is actually simpler than germ line edition, as it modifies the DNA sequence of only specific groups of cells, not of the entire individual. The exact effects of embryonic gene modification procedures are difficult to predict, and often become evident only after birth, in many cases, only a long time afterwards.
Fifteen European countries have enacted currently standing regulations forbidding the modification of the germ line, and the Recombinant DNA Advisory Committee from NIH has explicitly stated that it “will not entertain proposals for germ line alteration”.
In December 2015, Jennifer Doudna, a researcher at the University of California in Berkeley and one of the pioneers of the CRISPR/Cas9 technology, declared that “we do not yet know enough about the capabilities and limits of the new technologies, especially when it comes to creating heritable mutations … human-germ line editing for the purposes of creating genome-modified humans should not proceed at this time, partly because of the unknown social consequences, but also because the technology and our knowledge of the human genome are simply not ready to do so safely” [66]. Shortly thereafter, an international meeting took place, sponsored by the National Academy of Sciences of the USA, the National Academy of Medicine, the British Royal Society and the Chinese Academy of Sciences, to discuss the scientific and social implications of genome editing. The meeting made a call for a careful and prudent approach to research on heritable modifications of the human genome [67]. While acknowledging the potential of this research for the future eradication of genetic diseases and the improvement of human capabilities, it also declared that available technologies are still far below the standard required to do so with reasonable guarantees of success and safety. It was argued, however, that using germ cells or human embryos for basic and preclinical laboratory research was acceptable as long as the results were not intended for implantation. Not all attendants agreed though, and dissenting voices have been heard since.
In the United Kingdom, the Human Fertilisation and Embryology Authority (HFEA) has already approved experiments of embryonic gene editing as long as they use leftover embryos from in vitro fertilization procedures (February 2016) [68].
On August 2, 2017, the researchers Shoukhrat Mitalipov, Juan Carlos Izpisúa and Jin-Soo Kim performed the first experiments using CRISPR/Cas-based gene editing of human embryos in the USA, under the supervision of a bioethics committee assembled by the National Academy of Sciences of the USA [69]. Although this research goes against the agreements signed in Oviedo by 29 countries, including Spain, it should be noted that neither the USA, nor China or the United Kingdom are among the signees.
Ethical considerations regarding these procedures have polarized deeply the bioethics community, pitting those who think that all kinds of manipulation of the human embryo are immoral against those who see no reason for any ban. In the end, the positions of those adopting a more conciliatory approach (those who see no reason for an absolute prohibition, but demand that all human beings, even in the embryo stage, be treated with the utmost consideration and respect, therefore requesting prudence and assurances regarding the safety and efficacy of gene editing procedures) will prevail [70].
CONCLUSIONS
Although only a few years have passed since the discovery of the CRISPR/Cas system and its application in genetic engineering, this system is quickly becoming the premier tools for gene modification, enabling not only the quick and efficient implementation of genetic edits at the genomic level, but the specific and simultaneous modification of several genetic loci.
This system enables the introduction of point mutations, changes of larger sequences stretches by homologous recombination, the activation or repression of specific genes, the typing of bacterial strains, and the development of animal models for human diseases. There is virtually no end in sight for the number of future applications of this system, spanning fields as diverse as crop development, microbiological research, the food industry, drug development, and therapeutic applications in medicine.
While the versatility of the system provides a glimmer of hope regarding long-intractable problems, a healthy dose of realism must be used, considering that the application and development of CRISPR/ Cas technologies is still on its infancy, and that its most transcendent applications require considerable improvement of their efficacy and safety. Only when the benefits can be safely estimated to compensate for any potential hazards (as is the rule for any re-search on human beings) will we be able to leave behind basic research to enter the realm of therapeutic applications.
REFERENCES
1. Silva G, Poirot L, Galetto R, Smith J, Montoya G, Duchateau P, et al. Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr Gene Ther. 2011;11(1):11-27.
2. Jankele R, Svoboda P. TAL effectors: tools for DNA targeting. Brief Funct Genomics. 2014;13(5):409-19.
3. Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol. 1987;169(12):5429-33.
4. Nakata A, Amemura M, Makino K. Unusual nucleotide arrangement with repeated sequences in the Escherichia coli K-12 chromosome. J Bacteriol. 1989;171(6):3553-6.
5. Mojica FJ, Juez G, Rodriguez-Valera F. Transcription at different salinities of Haloferax mediterranei sequences adjacent to partially modified PstI sites. Mol Microbiol. 1993;9(3):613-21.
6. Mojica FJ, Ferrer C, Juez G, Rodriguez- Valera F. Long stretches of short tandem repeats are present in the largest replicons of the Archaea Haloferax mediterranei and Haloferax volcanii and could be involved in replicon partitioning. Mol Microbiol. 1995;17(1):85-93.
7. Mojica FJ, Diez-Villasenor C, Soria E, Juez G. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol Microbiol. 2000;36(1):244-6.
8. Jansen R, Embden JD, Gaastra W, Schouls LM. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol. 2002;43(6):1565-75.
9. Tang TH, Bachellerie JP, Rozhdestvensky T, Bortolin ML, Huber H, Drungowski M, et al. Identification of 86 candidates for small non-messenger RNAs from the archaeon Archaeoglobus fulgidus. Proc Natl Acad Sci U S A. 2002;99(11):7536-41.
10. Jiang F, Doudna JA. The structural biology of CRISPR-Cas systems. Curr Opin Struct Biol. 2015;30:100-11.
11. Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60(2):174-82.
12. Pourcel C, Salvignol G, Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology. 2005;151(Pt 3):653-63.
13. Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151(Pt 8):2551-61.
14. Mojica FJM, Garret RA. Discovery and Seminal Developments in the CRISPR Field. In: Barrangou R, van der Oost J, eds. CRISPR-Cas Systems RNA-Mediated Adaptative. Immunity in Bacteria and Archaea. Dordrecht: Springer; 2013. p. 299.
15. Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471(7340):602-7.
16. Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9(6):467-77.
17. Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709-12.
18. Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321(5891):960-4.
19. Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156(5):935-49.
20. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial im-munity. Science. 2012;337(6096):816-21.
21. Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346(6213):1258096.
22. ternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507(7490):62-7.
23. Shao Y, Guan Y, Wang L, Qiu Z, Liu M, Chen Y, et al. CRISPR/Cas-mediated genome editing in the rat via direct injection of one-cell embryos. Nat Protoc. 2014;9(10):2493-512.
24. Sakuma T, Yamamoto T. CRISPR/Cas9: The Leading Edge of Genome Editing Technology. In: T. Yamamoto, ed. Targeted Genome editing using site-specific nucleases. Tokyo: Springer. 2015. p. 205.
25. Holkers M, Maggio I, Henriques SF, Janssen JM, Cathomen T, Goncalves MA. Adenoviral vector DNA for accurate genome editing with engineered nucleases. Nat Methods. 2014;11(10):1051-7.
26. Kabadi AM, Ousterout DG, Hilton IB, Gersbach CA. Multiplex CRISPR/ Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res. 2014;42(19):e147.
27. Abrahimi P, Chang WG, Kluger MS, Qyang Y, Tellides G, Saltzman WM, et al. Efficient gene disruption in cultured primary human endothelial cells by CRISPR/ Cas9. Circ Res. 2015;117(2):121-8.
28. Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32(4):347-55.
29. Gratz SJ, Cummings AM, Nguyen JN, Hamm DC, Donohue LK, Harrison MM, et al. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 2013;194(4):1029-35.
30. Addgene: CRISPR/Cas9 Guide [Internet]. Addgene.org. 2017 [cited 2017 Sept 24]. Available from: https://www.addgene.org/crispr/guide/
31. Dow LE. Modeling disease in vivo with CRISPR/Cas9. Trends Mol Med. 2015;21(10):609-21.
32. Vanoli F, Jasin M. Generation of chromosomal translocations that lead to conditional fusion protein expression using CRISPR-Cas9 and homology-directed repair. Methods. 2017;121-122:138-45.
33. Evers B, Jastrzebski K, Heijmans JP, Grernrum W, Beijersbergen RL, Bernards R. CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat Biotechnol. 2016;34(6):631-3.
34. Ma H, Tu LC, Naseri A, Huisman M, Zhang S, Grunwald D, et al. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat Biotechnol. 2016;34(5):528-30.
35. Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, et al. Editing DNA methylation in the mammalian genome. Cell. 2016;167(1):233-47 e17.
36. Fellmann C, Gowen BG, Lin PC, Doudna JA, Corn JE. Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat Rev Drug Discov. 2017;16(2):89-100.
37. Luo J. CRISPR/Cas9: From Genome engineering to cancer drug discovery. Trends Cancer. 2016;2(6):313-24.
38. Dow LE, Fisher J, O’Rourke KP, Muley A, Kastenhuber ER, Livshits G, et al. Inducible in vivo genome editing with CRISPR-Cas9. Nat Biotechnol. 2015;33(4):390-4.
39. Wang X, Cao C, Huang J, Yao J, Hai T, Zheng Q, et al. One-step generation of triple gene-targeted pigs using CRISPR/Cas9 system. Sci Rep. 2016;6:20620.
40. Chen Y, Zheng Y, Kang Y, Yang W, Niu Y, Guo X, et al. Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9. Hum Mol Genet. 2015;24(13):3764-74.
41. Zhou J, Shen B, Zhang W, Wang J, Yang J, Chen L, et al. One-step generation of different immunodeficient mice with multiple gene modifications by CRISPR/ Cas9 mediated genome engineering. Int J Biochem Cell Biol. 2014;46:49-55.
42. Pazhakh V, Clark S, Keightley MC, Lieschke GJ. A GCSFR/CSF3R zebrafish mutant models the persistent basal neutrophil deficiency of severe congenital neutropenia. Sci Rep. 2017;7:44455.
43. Strong A, Musunuru K. Genome editing in cardiovascular diseases. Nat Rev Cardiol. 2017;14(1):11-20.
44. Cyranoski D. CRISPR gene-editing tested in a person for the first time. Nature. 2016;539(7630):479.
45. Reardon S. First CRISPR clinical trial gets green light from US panel. Nature. 2016. doi: 10.1038/nature.2016.20137.
46. Yin C, Zhang T, Qu X, Zhang Y, Putatunda R, Xiao X, et al. In Vivo Excision of HIV-1 Provirus by saCas9 and Multiplex Single-Guide RNAs in Animal Models. Mol Ther. 2017;25(5):1168-86.
47. Ma H, Marti-Gutierrez N, Park SW, Wu J, Lee Y, Suzuki K, et al. Correction of a pathogenic gene mutation in human embryos. Nature. 2017;548(7668):413-9.
48. Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24(1):132-41.
49. Ma Y, Zhang L, Huang X. Genome modification by CRISPR/Cas9. FEBS J. 2014;281(23):5186-93.
50. Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014;15(5):321-34.
51. Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827-32.
52. Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32(3):279-84.
53. Li K, Wang G, Andersen T, Zhou P, Pu WT. Optimization of genome engineering approaches with the CRISPR/Cas9 system. PLoS One. 2014;9(8):e105779.
54. Cong L, Zhang F. Genome Engineering Using CRISPR-Cas9 System. In: Pruett- Miller SM. Chromosomal Mutagenesis, Methods in Molecular Biology. New York: Springer; 2015. p. 197-217.
55. Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154(6):1380-9.
56. Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol. 2014;32(6):577-82.
57. Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529(7587):490-5.
58. Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods. 2013;10(10):957-63.
59. Viedma, I. Proyecto Genoma Humano. Implicaciones éticas. Persona y bioética. 2002;6(17):73-84.
60. Convenio Europeo sobre los derechos humanos y la biomedicina: Convenio para la protección de los derechos humanos y la dignidad del ser humano con respecto a las aplicaciones de la Biología y la Medicina. Convenio de Oviedo. Boletín Oficial de Estado. 14 de abril de 1997.
61. Declaración Universal sobre el Genoma Humano y los Derechos Humanos. UNESCO, París, 11 de Noviembre de 1997.
62. Gantz VM, Jasinskiene N, Tatarenkova O, Fazekas A, Macias VM, Bier E, et al. Highly efficient Cas9-mediated gene drive for population modification of the malaria vector mosquito Anopheles stephensi. Proc Natl Acad Sci U S A. 2015;112(49):E6736-43.
63. Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z, et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell. 2015;6(5):363-72.
64. Lanphier E, Urnov F, Haecker SE, Werner M, Smolenski J. Don’t edit the human germ line. Nature. 2015;519(7544):410-1.
65. Baltimore D, Berg P, Botchan M, Carroll D, Charo RA, Church G, et al. Biotechnology. A prudent path forward for genomic engineering and germline gene modification. Science. 2015;348(6230):36-8.
66. Doudna J. Perspective: Embryo editing needs scrutiny. Nature. 2015;528(7580):S6.
67. Belluz J. Why scientists are calling for caution on a powerful new gene-editing technology. Vox Science & Health. 2015 December 3. [cited 2017 Sept 24]. Available from: http://www.vox.com/2015/12/3/9845230/crispr-gene-editing-caution
68. Licence Committee HFEA. The Human Fertilisation and Embryology Authority (HFEA). 2016 Feb. Available from: https://www.hfea.gov.uk/about-us/our-committees-and-panels/scientific-and-clinical-advances-advisory-committee-scaac/
69. National Academies of Sciences, Engineering, and Medicine. Human Genome Editing: Science, Ethics, and Governance. Washington, DC. 2017.
70. Bueren JA, Gracia D. Terapia génica en línea germinal: Aspectos científicos y éticos. In: Ayuso C, Dal-Ré R, Palau F. Ética de la investigación de las enfermedades raras. Madrid: Ed. Ergon; 2016. p. 147-62.
Received in May, 2017.
Accepted in November, 2017.
Inmaculada Viedma. Facultad de Enfermería de la Universidad Católica de Murcia, España. E-mail: iviedma@ucam.edu.


{kind=link}
{kind=link}