










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Plantas Medicinales
versión On-line ISSN 1028-4796
Rev Cubana Plant Med v.16 n.1 Ciudad de la Habana ene.-mar. 2011
ARTÍCULO ORIGINAL
Validación del método de cuantificación de taninos totales en formulaciones semisólidas de Rhizophora mangle L. (mangle rojo)
Validation of the total tannin quantitative determination method in Rhizophora mangle L. (red mangrove) semi-solid formulations
María del Carmen Travieso Novelles,I Arsenio Betancourt Bravo,II Arturo Escobar Medina,III Anel Linares Nuñez,IV Yanet Rodríguez Perdomo,V Tania Pérez BuenoVI
I Licenciada en Ciencias Farmacéuticas. Doctora en Ciencias. Investigadora Auxiliar. Centro Nacional de Sanidad Agropecuaria (CENSA). La Habana, Cuba.
II Licenciado en Bioquímica. Doctor en Ciencias. Investigador Auxiliar. CENSA. La Habana, Cuba.
III Licenciado en Química. Doctor en Ciencias. Investigador Titular. CENSA. La Habana, Cuba.
IV Ingeniera Química. Reserva Científica. CENSA. La Habana, Cuba.
V Licenciada en Radioquímica. CENSA. La Habana, Cuba.
VI Ingeniera Química. Doctora en Ciencias. Investigadora Auxiliar. CENSA. La Habana, Cuba.
RESUMEN
Introducción: extractos acuosos de Rhizophora mangle L. (mangle rojo) presentan alto contenido de taninos que le confieren propiedades de antiséptico y cicatrizante.
Objetivo: demostrar la validez de un método de cuantificación de taninos totales para el control de calidad de formulaciones semisólidas de mangle rojo.
Métodos: se estudiaron los indicadores de linealidad, precisión, exactitud, selectividad, y robustez, sugeridos por la USP y la Regulación 41 del CECMED para métodos cuantitativos de contenido o pureza de principios activos mayoritarios.
Resultados: se demostró la linealidad en el rango de 0,40-1 mg/mL del patrón empleado, se obtuvo la ecuación de la recta, con coeficientes de determinación y correlación (r2 y r) de 0,9912 y 0,9956; el intercepto y la pendiente, no difirieron 0 y 1, respectivamente. Los estudios de recobrado mostraron porcentajes de recobro superiores a 98 % en los diferentes niveles de concentración ensayados indicativos de una adecuada exactitud. El método resultó ser preciso tanto en condiciones óptimas de repetibilidad como en condiciones intermedias. Los resultados de la selectividad mostraron que la respuesta del método no presentó interferencias con otros componentes de la formulación, así como la ausencia de respuesta en los placebos. La robustez quedó comprobada al no obtenerse diferencias significativas entre los resultados obtenidos de corridas del ensayo normal y corridas con cambios (temperatura y tiempo de la centrifugación, variaciones pequeñas del pH del buffer) que resultaron no significativos.
Conclusiones: el método de cuantificación de taninos totales resulta confiable para la evaluación de estos compuestos en la formulación semisólida estudiada.
Palabras clave: Rhizophora mangle L., validación, formulaciones semisólidas, taninos totales.
ABSTRACT
Introduction: Rhizophora mangle L. (red mangrove) aqueous extracts have high tannin content that gives them antiseptic and healing features.
Objective: to demonstrate the validity of total tannin quantitative determination method for the quality control of semi-solid red mangrove formulations.
Methods: The linearity, precision, accuracy, selectivity and robustness indicators were studied in pursuant to the USP guidelines and the Regulation 41 of CECMED for the quantitative determination methods of the content or purity of majority active principles.
Results: the linearity in the 0.40-1 mg/mL range of the used pattern was proved, the equation of the straight line with determination and correlation (r2 y r) coefficients equal to 0.9912 and 0.9956 respectively were obtained, and the intercept and the slope did not differ from 0 and 1, respectively. The recovery studies showed recovery rates over 98 % at the various levels of concentration tested, which indicated adequate accuracy. The method was precise both in optimal repeatability conditions and intermediate conditions. The selectivity results revealed that the method response did not interfere with other components of the formulation, and that there was no response in the placebos. Robustness was confirmed since no significant differences between the results from the normal test runs and from the test runs including changes (temperature and centrifugation time, slight variations in the buffer pH) were observed, being all these results not significant.
Conclusions: the total tannin quantitative determination method was reliable for the evaluation of these compounds in the studied semi-solid formulation.
Key words: Rhizophora mangle L., validation, semi-solid formulations, total tannins.
INTRODUCCION
El desarrollo de medicamentos de fuentes naturales constituye una prioridad en el mundo, debido a la eficacia demostrada por los principios activos obtenidos de estas fuentes, así como otras ventajas relacionadas con la disminución de las reacciones adversas y toxicidades asociadas, además de la disponibilidad de las materias primas vegetales si se sustentan en un programa de explotación sostenible.1 Los organismos de salud (OMS) internacionales estimulan, cada vez más, la investigación y búsqueda de fármacos a partir de fuentes naturales, basados en la experiencia tradicional del uso de estas fuentes para aliviar y curar enfermedades y exigen el inventario sistemático de los ensayos preclínicos y clínicos que se realizan con productos derivados de plantas.2 Dentro de la diversa y abundante flora tropical, a partir de la cual se han obtenido nuevos candidatos a productos naturales de actividad probada en estudios in vivo e in vitro,3 desde hace algunos años se investiga en la evaluación farmacológica y el desarrollo de formulaciones a partir de Rhizophora mangle L., para uso animal y humano, con resultados preclínicos y clínicos que han demostrado su actividad y eficacia como cicatrizante, antiséptico y antioxidante,4-7 debido mayoritariamente a la presencia de taninos. Por ser estos compuestos fenólicos (taninos), los principios activos de estas preparaciones, ha sido necesario contar con un método de análisis cuantitativo confiable para ser empleado, tanto en la investigación como en los controles de proceso de los productos intermedios y en el control de calidad de los productos finales. Por esta razón, se estandarizó un método basado en la precipitación con proteínas de los compuestos fenólicos sugerido por Hagerman y Butler,8 y posteriormente validado para su uso en la evaluación de formulaciones líquidas.9 Hoy día se desarrollan otras formulaciones semisólidas para mejorar la biodisponibilidad por vía tópica. El presente trabajo enfoca los estudios de validación de este método analítico en nuevas formulaciones semisólidas obtenidas a partir de extracto acuoso de mangle rojo, con vistas a demostrar su validez para emitir resultados confiables.
MÉTODOS
Se utilizaron albúmina sérica bovina (BSA), laurilsufato de sodio, trietanolamina, cloruro férrico, ácido tánico, cloruro de sodio, acetato de amonio y ácido clorhídrico, todos de la Merck. Los equipos de medición empleados fueron: balanza analítica (Mettler H35 AR), balanza digital (Sartorius Portable), espectrofotómetro (Ultrospec Plus pharmacia LKB), espectrofotómetro (Lasso Biotech LTDA), peachímetro (Autocal 83). El resto de los equipos están declarados aptos para el uso: centrífuga refrigerada (Jouan KR 4-22), agitador de tubos (VF-2). La cristalería de medición exacta utilizada fue declarada apta para el uso y las pipetas automáticas estuvieron debidamente calibradas.
Se emplearon soluciones de albúmina sérica bovina (1 mg/mL) en buffer de acetato de amonio 0,2 M a pH 5,0 que contenía cloruro de sodio 0,7 M; solución de laurilsulfato de sodio 1 % p/v, solución de laurisulfato de sodio (SDS) 1 % y trietanolamina 7 %, solución de cloruro férrico (FeCL3 0,01 M en HCl 0,1 N).
Se utilizó como estándar ácido tánico. La curva de calibración se preparó de una solución madre de 1 mg/mL, a partir de la cual se hicieron diferentes niveles de concentraciones: 0,4; 0,6; y 0,8 mg/mL.
Preparación de las muestras
Consistió en solubilizar la muestra problema, se pesó 1 g del semisólido que se transfirió totalmente a un volumétrico con agua destilada. En general se utilizaron diluciones de 1:25 o 1:50 en dependencia de la concentración esperada de taninos.
La marcha analítica resumida consistió en la toma, en cada tubo de ensayo, de una alícuota de 1 mL de cada punto de la curva y de la muestra problema y la adición de 2 mL de la solución estándar de proteínas (BSA). Se mezclaron las soluciones y se dejaron reposar a temperatura ambiente durante 15 min. Posteriormente se centrifugaron durante 15 min a 2 500 rpm. Se lavó la superficie del residuo y las paredes del tubo sin destruir el pellet con el buffer acetato; se decantó este lavado y se disolvió el pellet en 1,5 mL de la solución laurilsulfato de sodio 1 %, agitando fuertemente. Después se tomó una alícuota de 1 mL de cada solución (patrones y muestras) y se añadieron 3 mL de la solución de laurilsulfato de sodio, trietanolamina y 1 mL de cloruro férrico. Se agitó de nuevo alrededor de 15 s y se leyó la densidad óptica a 510 nm después de transcurrido de 15 a 30 min.
Metodología de la validación
Este método pertenece a la categoría I (Manual de la FDA y USP 30) de métodos cuantitativos para evaluar componentes mayoritario, por lo que fue necesario la demostración de los indicadores: linealidad, precisión, exactitud, robustez, y especificidad/selectividad.10,11
La linealidad se evaluó mediante curvas de calibración, para lo cual se empleó como patrón el ácido tánico. Se prepararon 5 niveles de concentración en un rango de 30 a 120 % del valor esperado. Se partió de una solución madre de 1 mg/mL, a partir de la cual se prepararon los restantes puntos de la curva, se realizaron 5 repeticiones por cada nivel de concentración teniendo en cuenta la marcha analítica descrita. Se hizo un análisis de regresión lineal, mediante el sistema Excel, y se determinó la recta de mejor ajuste, los coeficientes de correlación (r) y de determinación (r2), así como la pendiente y el intercepto; se tomó como criterios de una adecuada linealidad cuando r > 0,99; r2 > 0,98; y que la pendiente y el intercepto no difiriera de 1 y 0, respectivamente.
La exactitud se evaluó mediante estudio de recobrado. Se prepararon diferentes niveles de concentraciones a partir de una muestra base, mediante la adición de concentraciones conocidas de patrón. Estas muestras se analizaron en paralelo por quintuplicado y se cuantificaron mediante curva de calibración preparada con soluciones de patrones. Se calculó la recuperación como medida de exactitud y su significación por pruebas estadísticas apropiadas.
Se preparó un nivel base y 6 niveles de concentración que estuvieron dentro del rango de la curva de calibración. Para cada porción de ensayo se pesó 1 g del semisólido en balanza analítica y se adicionó a un beaker pequeño. La masa se trasvasó completamente a un volumétrico de 50 mL poco a poco arrastrando todo el contenido pesado. A continuación se adicionaron las concentraciones conocidas de solución patrón de ácido tánico (1 mg/mL) para lograr concentraciones añadidas en el rango de 0,04 - 0,4 mg/mL. Se enrazó con agua desionizada hasta 50 mL. Se realizó una curva de recuperación con los niveles antes mencionados, con 5 repeticiones por cada nivel. Se determinaron las concentraciones observadas según la marcha analítica descrita antes; y las concentraciones esperadas y los porcentajes de recuperación se calcularon aplicando las ecuaciones (I) y (II).

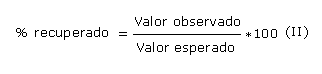
Se realizó análisis estadístico, mediante el sistema Excel y se comprobó la relación entre las concentraciones observadas y esperadas, y se determinó el coeficiente de correlación (r, que debe mostrar valores superiores a 0,99 como indicativo de una adecuada exactitud.
Los estudios de precisión se realizaron tanto en condiciones óptimas (repetibilidad), y en condiciones intermedias. Se demostró la repetibilidad evaluando la variabilidad de los resultados en tres niveles de concentraciones (alto, medio y bajo) de taninos totales (mg/mL), para lo cual se pesaron 1,5; 1; y 0,5 g de un mismo lote de producto, los cuales se trasvasaron íntegramente a volumétricos de 50 mL. Se hicieron 5 repeticiones por cada nivel de concentración siguiendo la marcha descrita bajo condiciones óptimas (analista, día, equipo, y lote de reactivos idénticos). Se calculó la desviación estándar de repetibilidad (Sr) y el intervalo crítico (CR), mediante las ecuaciones (III) y (IV).12

Donde:
t: número de niveles.
n: repeticiones por cada nivel.
Yjk: resultado de la prueba para cada réplica.
Y: valor medio de los resultados de n réplicas.
Se determinó el coeficiente de variación, el cual no debe ser superior a 3 % (métodos espectrofotométricos).
CR (n)= f(n) * Sr (IV)
Donde:
f(n): factor tabulado de intervalo crítico para una n determinada (para n=3, f [n]= 3,3)
n: número de replicas en cada determinación.
Para el ensayo de precisión en condiciones intermedias se trabajó con los mismos niveles de concentración que en el ensayo de repetibilidad.
En este punto se evaluó la variabilidad de los resultados bajo la influencia de diversas variables, teniendo en cuenta diferentes situaciones como: operador (O), tiempo (T), lote de reactivo color (R) y equipo (E), así como algunas interacciones entre estas variables, lo cual permitió calcular la desviación estándar de precisión en condiciones intermedias (Si), para cada una de estas situaciones.
Situaciones evaluadas según necesidades prácticas:
- 2 analistas, un mismo día Si (O).
- 1 analista, 2 d diferentes, con lotes de reactivos diferentes Si (T, R).
Se realizaron 5 repeticiones por cada nivel de concentración en cada situación evaluada, siguiendo el mismo procedimiento de análisis anterior.
Se calculó la desviación estándar de la precisión en condiciones intermedias (Si), para cada una de las condiciones propuestas según (III), y se determinó el intervalo crítico (CR), mediante (IV).
Para evaluar la selectividad se emplearon muestras del mismo lote y placebo. Se evaluó la capacidad del método para detectar los productos de interés (taninos) de forma diferenciada de otros compuestos que pudieran estar presentes en la matriz (excipientes), para lo cual se evaluaron comparativamente 3 muestras contra placebo. Se realizaron 5 réplicas por cada muestra ensayada. Después se compararon los resultados mediante una prueba no paramétrica para n pequeña (Wilcoxon), que permite de esta forma comprobar si existen diferencias significativas entre las muestras.
Para la demostración de la robustez se evaluaron variaciones razonables efectuadas con el propósito de identificar factores que pudieran originar variaciones menores o significativas en el método de ensayo. Las variaciones se realizaron dentro de la misma corrida con vistas a efectuar la estimación del efecto global de cada factor.
Se evaluó de forma simultánea la influencia de:
- La temperatura durante la centrifugación (temperaturas de 27 y 35 °C).
- Tiempo de centrifugación: 10 y 20 min.
- pH del buffer acetato: 5; y 5,5.
- Con lavado y sin lavado.
Se compararon estadísticamente los resultados de las corridas de ensayos realizadas según el procedimiento con los obtenidos tras realizar los cambios simultáneos, para evaluar y clasificar los cambios en significativos y no significativos.
RESULTADOS
Al procesar estadísticamente los resultados del estudio de linealidad se comprobó el comportamiento lineal del sistema; se observó que existe una correlación entre la densidad óptica y la concentración en el rango estudiado, se obtuvo la ecuación de la recta Y= 1,365 X-0,0045, donde los coeficientes de determinación y correlación (r2 y r) fueron de 0,9912 y 0,9956, respectivamente; el intercepto no difirió significativamente de 0 ni la pendiente de 1.
Los resultados del estudio de exactitud (tabla 1), demuestran que el procedimiento en estudio es exacto, puesto que los porcentajes de recobrado obtenidos se encuentran en el rango establecido para este tipo de método (> 80 %). Al analizar la relación existente entre las concentraciones esperadas y observadas, se obtuvo una línea recta cuyo coeficiente de correlación resultó superior a 0,99; lo cual corrobora la exactitud del método.

El estudio de precisión realizado bajo condiciones de repetibilidad (tabla 2), permitió conocer la Sr y el CR bajo condiciones óptimas (analista, equipo, reactivo, día idéntico). Se obtuvieron resultados similares para las 4 repeticiones de este estudio. De esa forma se pudo conocer la máxima diferencia permisible entre réplicas, primer factor a tener en cuenta por el analista para conocer sobre la validez del ensayo.
El procesamiento estadístico de los resultados del ensayo de precisión en condiciones intermedias (tabla 2), muestra que no existen diferencias significativas (p< 0,05), al aplicar la prueba t, entre las diferentes condiciones intermedias analizadas; sin embargo, se observó aumento de la variabilidad con respecto a las condiciones de repetibilidad, evidenciado además por los valores de rangos críticos que casi se duplicaron.
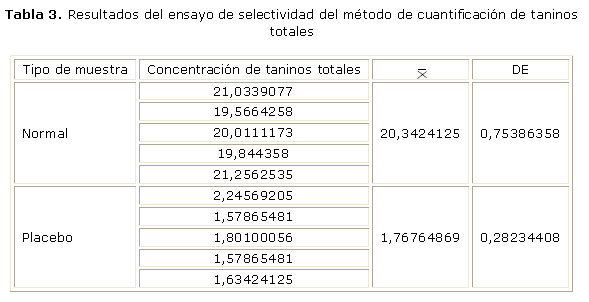
La selectividad del método quedó demostrada al no observarse respuesta del placebo (tabla 3), lo cual evidencia que el resto de los componentes de la formulación no provocan interferencias.
Los resultados del estudio de la robustez mostraron que el método no es influenciable por ninguna de las fuentes ensayadas (temperatura y duración de la centrifugación, así como variaciones pequeñas del pH del buffer), lo cual informa que estos no son cambios significativos porque no afectaron estadísticamente los resultados (tabla 4), lo que permitió demostrar la robustez del método.
DISCUSIÓN
Los resultados del estudio de linealidad demostraron la capacidad de este método de asegurar que los resultados analíticos obtenidos son proporcionales a la concentración del analito dentro del intervalo de 0,4-1,00 mg/mL del patrón empleado13,14. Esto coincide con autores que validaron otro método espectrofotométrico de cuantificación de taninos totales y reportan similares coeficientes de determinación y correlación.15 Estos resultados demuestran que el método es lineal en el rango de concentraciones de la curva patrón, se muestra de esta forma que el ensayo cumple de modo apropiado con la Ley de Lambert-Beer, lo cual indica que se pueda usar la absorbancia como magnitud adecuada para determinar a partir de ella la concentración de principio activo presente en las muestras.16
Por otra parte, los resultados de la exactitud mostraron altos niveles de recobro,17 además de coincidir con resultados anteriores de este método en otras formulaciones farmacéuticas.9 Algunos autores plantean la aceptación de valores de recobrado no menores que 50, 80 y 90 %, como límites de aceptación numéricos en dependencia de la complejidad del método.18,19 Otros autores refieren altos recobrados para métodos espectrofotométricos simples.20 Los valores de recobrado obtenidos son similares a los alcanzados por otros autores que reportan valores de porcentajes de recobro superiores a 97 % en un método de cuantificación de estos compuestos.15
El estudio de precisión mostró un aumento de la variabilidad de los resultados cuando se trabaja en condiciones no óptimas; se duplican prácticamente los valores en condiciones intermedias, lo cual coincide con lo explicado en la ISO 5725 (3), referente al aumento de la variabilidad de los resultados cuando se realiza el método en condiciones intermedias de precisión.12 Por otra parte, el análisis de los coeficientes de variación mostró la influencia del cambio de analista en la variabilidad de los resultados, así como la influencia del día de análisis y el lote de reactivo color. Estos resultados avalan la importancia de tener en cuenta, en este tipo de ensayo, factores técnicos como el adiestramiento del personal, entre otros factores críticos.
Otros resultados fueron la demostración, en el estudio de selectividad, de la no interferencia de estos nuevos componentes de las formulaciones, que asegura el hecho de estar determinando exclusivamente la sustancia de interés;21,22 así como la robustez puesto que los resultados demostraron que las respuestas obtenidas mediante la ejecución del ensayo, no son afectados por pequeñas variaciones en las condiciones de trabajo.21
Se puede concluir que el método de cuantificación de taninos totales resulta adecuado para la evaluación de estos compuestos en la formulación semisólida utilizada, porque resultó preciso, exacto, selectivo, robusto y lineal.
AGRADECIMIENTOS
Constituye un principio para la validez de los estudios de validación de los métodos analíticos, que estos sean llevados a cabo por el personal que rutinariamente ejecuta estos ensayos con alto grado de confiabilidad, por lo que agradecemos por su contribución en la ejecución de los diseños experimentales de este trabajo, a las analistas principales de esta línea de investigación Daysi González y Gisel Vibo.
REFERENCIAS BIBLIOGRÁFICAS
1. OMS. Directrices de la OMS sobre buenas prácticas agrícolas y de recolección (BPAR) de plantas medicinales. ISBN 978-924-354-627-8. Ginebra; 2003. p. 86.
2. OMS. Situación reglamentaria de los medicamentos herbarios: una reseña mundial. Ginebra, OMS/TRM; 2000. p. 60.
3. Salomón SI, López OH, García CM, González MS, Fusté VM. Desarrollo de una tecnología para la obtención de extracto acuoso de hojas de Morinda citrofolia L. Rev Cubana Plant Med. 2009;14(2):1-8.
4. Armenteros M. Evaluación de un desinfectante mamario post-ordeño de origen natural [Tesis doctoral]. La Habana: Centro Nacional de Sanidad Agropecuaria-Universidad Agraria de La Habana; 1998.
5. Sánchez LM. Caracterización química y actividad biológica de un extracto acuoso de la corteza de Rhizophora mangle L. [Tesis doctoral]. La Habana: Centro Nacional de Sanidad Agropecuaria-Universidad Agraria de La Habana; 1998.
6. Melchor G. Efectos cicatrizante y antiséptico del extracto y de una forma farmacéutica obtenida a partir de Rhizophora mangle L. [Tesis doctoral]. La Habana: Centro Nacional de Sanidad Agropecuaria-Universidad Agraria de La Habana; 1999.
7. Sánchez JC. Propiedades antioxidantes del extracto acuoso de Rhizophora mangle L. y de su fracción polifenólica mayoritaria evaluadas en sistemas in vitro e in vivo. [Tesis doctoral]. La Habana: Centro Nacional de Sanidad Agropecuaria- Universidad Agraria de La Habana; 2007.
8. Hagerman AE, Butler LG. Protein precipitation method for the quantitative determination of tannins. J Agricultural Food Chemistry. 1978;26(4):809-12.
9. Travieso MC, Hechavarría OL, Betancourt AB, Frontera M, Miranda I. Validación del método de determinación de taninos totales en productos naturales vegetales. Rev Protección Veg. 1998;13(3):189-94.
10. Food and Drug Administration. Manual de Laboratorio FDA ORA. Section 2. Technical Requirements. Methods, method verification and validation. ORA-LAB 5.4.5; 2004.
11. United State Pharmacopeia. General Chapter 1225. Validation of compendial methods. Ed. 30. National Formulary. Rockville Md USA: Pharmacopeia Convention Editorial; 2007. p. 25.
12. ISO-5725. Precision of test methods. Determination of repeteability and reproducibility for a standard test method by inter-laboratory test. Part 3; 1994.
13. Gil AE. Protocolo de validación de métodos de análisis. Materias primas y productos terminados. Departamento de Farmacia y Tecnología Farmacéutica. Facultad de Farmacia. España: Universidad Complutense de Madrid; 1995.
14. Gonzáles R. Protocolo de Validación de Técnicas Analíticas para la determinación cuantitativa en el control de la calidad y/o en estudios de estabilidad de materias primas o principios activos de medicamentos. Cuba: Centro de Química Farmacéutica. División de calidad; 1996.
15. Gutiérrez GY, Miranda MM, Varona TN, Rodríguez AT. Validación de dos métodos espectrofotométricos para la cuantificación de taninos y flavonoides (Quercetina) en Psidium Guajaba L. Rev Cubana Farm. 2000;34(1):50-5.
16. Massart DL, Verbeke JS. An introduction to method validation. Analusis Magazine. 1994;2(5).
17. Thomas H, Gerald S. Validation of Bioanalytical Methods. Pharmaceutical Research. 1991;8(4):421-6.
18. Castro N, Gascón S, Pujol M, Sans JM, Vicent L. Validación de métodos analíticos. Asociación Española de Farmacéuticos de la Industria. Monografía AEFI. Sección Catalana. Comisión de Normas de Buena Fabricación y Control de la Calidad. España: Edición Hewlett Packard; 1989. p. 19-42.
19. Campmany AC. Validación de métodos analíticos. Farm Clin. 1990;7(9):749-58.
20. Rodríguez AM, Blanco MR, Almiral ID. Desarrollo y validación de un método analítico espectrofotométrico para la cuantificación de tabletas de ibuprofeno 200 mg. Anuario Científico CECMED. 2003;1(Sup Esp):91-8.
21. Pasteelnik LA. Analytical methods validation. In: Berry R, Robert NA, editors. Pharmaceutical process validation. 2nd ed. New York: Ed. Marcell Dekker; 1993. p. 411-28.
22. Tanase IG, Popa DE, Buleandra M. Validation of an analytical quantitative determination method of chloride anion from drinking and surface water, using direct potentiometry with chloride- selective electrode (II). Anal University of Bucharest. Chimie Anul. 2007;XVI(II):1-57.
Recibido: 29 de marzo de 2010.
Aprobado: 30 de diciembre de 2010.
María del Carmen Travieso Novelles. Centro Nacional de Sanidad Agropecuaria Carretera de Tapaste y Ocho Vías, San José de las Lajas. AP 10. La Habana, Cuba. Correo electrónico: mcarmen@censa.edu.cu


{kind=link}
{kind=link}
{kind=link}