










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Plantas Medicinales
versión On-line ISSN 1028-4796
Rev Cubana Plant Med vol.19 no.3 Ciudad de la Habana jul.-set. 2014
ARTÍCULO ORIGINAL
Validación de una técnica por Cromatografía Líquida de Alta Resolución
para la determinación del contenido de Mangiferina en hojas de Mangifera indica L
Validation of a High Performance Liquid Chromatography technique for determination of mangiferin content in Mangifera indica L. leaves
Lic. Jacqueline Aylema Romero Díaz,I DrC. Lauro Nuva-Paz,II Téc. Marilin López,I Téc. Carlos Ferrada,I Téc. Caridad CarballoI
I Centro de Investigación y Desarrollo de Medicamentos (CIDEM). La Habana, Cuba.
II Centro Nacional de Genética Médica. La Habana, Cuba.
RESUMEN
Introducción: la Mangiferina es una glicosilxantona natural presente en varias partes del árbol de Mangifera indica L., a la que se le atribuyen un amplio rango de acciones farmacológicas.
Objetivo: establecer un método de determinación del contenido de Mangiferina en las hojas de Mangifera Indica L. por Cromatografia Líquida de Alta Resolución.
Métodos: para cuantificar el metabolito en las hojas, previamente secadas y pulverizadas, la separación se realizó a través de una columna cromatográfica Luna RP-18 (5 m) (250 x 4 mm), con detector UV a 254 nm usando un gradiente de fase móvil A compuesta de Acético ácido 0,2 %: acetonitrilo (85:15) y móvil fase B: Acetonitrilo y la cuantificación de este frente a una muestra de referencia utilizando el método del estándar externo.
Resultados: la ecuación de la curva de regresión de Mangiferina fue Y = 3,5561 X + 6536,55 (r = 0,999) El porciento de recobrado fue de 99,41 con un coeficiente de variación inferior al 2 %. Los límites de detección y cuantificación fueron de 0.0009 y 0,001 respectivamente. No se observaron diferencias estadísticamente significativas entre las determinaciones realizadas en diferentes días por los diferentes analistas.
Conclusión: El método es simple y fiable, el cual puede ser utilizado para el control de la calidad y estudio de estabilidad de las hojas de Mangifera Indica L.
Palabras clave: Mangiferina, CLAR, validación.
ABSTRACT
Introduction: Mangiferin is a natural glycosylxanthone present in various parts of the tree Mangifera indica L. which is attributed a wide range of pharmacological properties.
Objective: establish a method to determine the content of mangiferin in Mangifera Indica L. leaves by high performance liquid chromatography.
Methods: leaves were dried and pulverized before quantification of their metabolite content. Separation was performed using a Luna RP-18 chromatographic column (5 µm) (250 x 4mm) with a UV detector at 254n and a mobile phase A gradient composed of acetic acid 0.2 %: acetonitrile (85:15), and a phase B mobile: acetonitrile and its quantification vs. a reference sample applying the external standard method.
Results: the regression curve equation for mangiferin was Y=3.5561X +6536.55(r=0.999). Recovery was 99.41 % with a variation coefficient below 2 %. Detection and quantification limits were 0.0009 and 0.001, respectively. No statistically significant differences were found between the determinations performed on different days by different analysts.
Conclusion: the method is simple and reliable, and may be used for quality control and stability studies about Mangifera Indica L. leaves.
Key words: mangiferin, HPLC, validation.
INTRODUCCIÓN
La Mangifera indica L. es un árbol ampliamente distribuido en nuestro país perteneciente a la familia Anacardiáceas, conocida como planta alimenticia por sus frutos y como planta medicinal por sus propiedades entnomédicas.1
El uso popular del extracto acuoso de las diferentes partes de M. indica L. es ampliamente empleado en la medicina natural y tradicional por sus propiedades analgésico, antinflamatorio., inmunoestimulante, antioxidante; espasmolítico, antidiarreico, dislipidémico, antidiabético, antihelmíntico, antialérgico y antibacteriano.2-15
Estudios químicos reportados demuestran la presencia de polifenoles entre ellos la Mangiferina (2-β-D-glucopiranosil-1,3,6,7-tetrahidroxixanten-9-ona) la cual es una glucosilxantona natural que se desataca como el compuesto mayoritario y está presente en varias partes de M. indica: hojas, frutos, corteza, duramen y raíces.16-21
Además dicho compuesto ha sido caracterizado en las familias de las Anacardiáceas y Gentianáceae, especialmente en las hojas y la corteza.22
Estudios recientes indican que la Mangiferina (MgF) tiene un amplio rango de actividades farmacológicas, incluyendo las acciones antioxidantes, antidiabéticas, anti-VIH, antitumorales, hepatoprotectoras, antivirales, anticancerígenos, cardioprotectores e hipolipidémico.22-27
Existen reportados en la literatura métodos para la determinación de MgF, incluyendo la electroforesis capilar,28 cromatografía liquida acoplada a espectrometría de masas en tándem (LC-MS-MS),29 cromatografía líquida de alto rendimiento de fase reversa (RP-HPLC) con detección UV en preparaciones chinas,30 en plasma de rata y en la orina,31 mediante foto-colorimetría,32 métodos espectrofotométricos,33 y espectrofluorimétricos.34 El objetivo del presente trabajo fue validar un método analítico mediante cromatografía líquida de alta resolución para la determinación del contenido de MgF presente en las hojas de M. indica.
MÉTODOS
Recolección
El material vegetal se colectó en mayo de 2012, en la Estación Experimental de Plantas Medicinales “Dr. Juan Tomás Roig” en San Antonio de los Baños y la misma fue identificada por el Dr. Víctor R. Fuentes del Instituto de Investigaciones en Fruticultura Tropical y se conserva una muestra con el número de herbario ROIG 4776.
El material vegetal (hojas) fue desecado en un secador solar hasta obtener peso constante, se procedió a la pulverización de la droga en un molino de martillo hasta obtener un polvo grueso (0,5 mm) almacenándose en bolsas de Polietileno-Aluminio para su posterior uso en la elaboración de los extractos.
Reactivos
La sustancia de referencia química, con una pureza de 93,8 %, se encuentra registrada en el Centro Estatal para el Control de Medicamentos (CECMED), Cuba. Los reactivos utilizados fueron de calidad analítica.
Preparación de la sustancia de referencia
Para la construcción de la curva de calibración, se preparó una solución madre de 100 ppm con la MgF de referencia en una solución de Dioxano 50 %. A partir de ella, se prepararon patrones en agua de 2, 4, 8, 16 y 32 ppm, respectivamente.
Preparación de la muestra
Se pesó alrededor de 1g de hojas secas y se procedió a la elaboración de un extracto mediante extracción continua en un equipo Soxhlet, utilizando dioxano como solvente de extracción durante 4 horas. Se trasvasó el extracto a un matraz aforado de 500 mL y se completó a volumen con una mezcla Dioxano: Agua (1:1) v/v. Se tomó una alícuota de 5 mL y se llevó a un matraz aforado de 50 mL y se completó a volumen con la misma mezcla.
Determinación analítica
Se procedió empleándose la cromatografía líquida de alta resolución (HPLC), donde el sistema cromatográfico compuesto por una bomba Smartline-1000 conectada a un detector de longitud de onda variable Smartline-2500 (Knauer, Alemania.), con un volumen de inyección de 20-μL y la señal procesada por el software ClarityChrom versión 2.6.5.517. La fase móvil óptima consistió en una mezcla desgasificada de ácido acético 0,2 %: acetonitrilo (85:15) (V/V). Para la separación cromatográfica se utilizó una columna Luna, 5μm RP C18, 250 x 4,6 mm. Se estableció una longitud de onda de 254 nm en el detector de rayos ultravioletas. La elución fue llevada a cabo mediante el gradiente que se muestra en la (Tabla 1).
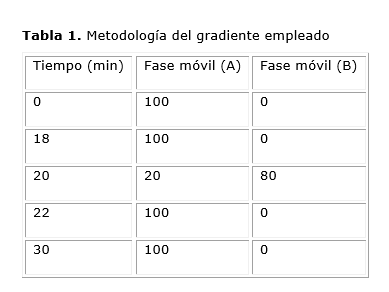
La cuantificación se realizó mediante una curva de calibración del material de referencia químico de MgF que abarcó un rango entre 2,0 y 50,0 µg/mL.
Se colectó manualmente el pico correspondiente al tiempo de retención de la MgF y se realizó un barrido desde 200 hasta 400 nm, utilizando un Espectrofotómetro GENESYS 2 (SPECTRONIC, USA)
Validación
Para la evaluación de la especificidad, los extractos fueron sometidos a condiciones drásticas como: hidrólisis ácida HCl 5 N, hidrólisis básica NaOH 1N, oxidación con peróxido de hidrógeno.
La precisión se llevó a cabo mediante la prueba de repetibilidad y la precisión intermedia. En el caso de la repetibilidad se realizaron 6 réplicas y para la precisión intermedia, 5 determinaciones por parte del primer analista y 5 determinaciones por parte del segundo analista en diferentes días, siguiendo el método de análisis propuesto con anterioridad. Se determinó la media, la desviación estándar y el coeficiente de variación para cada analista. Los resultados obtenidos se compararon estadísticamente mediante la prueba de Fisher y la prueba de t de Student, utilizando el programa estadístico Stargraphic plus versión 4,1, con vista a establecer si existían diferencias significativas entre los resultados.
La exactitud se evaluó mediante estudio de recobrado. Se prepararon diferentes niveles de concentraciones a partir de una muestra base, mediante la adición de concentraciones conocidas de patrón. Estas muestras se analizaron en paralelo por triplicado. Se determinaron los porcentajes de recuperación, las desviaciones estándar y los coeficientes de variación. Se determinaron además las pruebas de Cochram con vistas a comprobar si la variación de la concentración produce diferencias significativas en los resultados y las pruebas de la t de Student para la determinación de las diferencias significativas entre la recuperación media y el 100.
La linealidad del sistema cromatográfico se evaluó realizando 3 réplicas para 5 concentraciones diferentes que en el intervalo 2 - 100 µg/mL. Se determinaron las ecuaciones de las rectas, los coeficientes de correlación, las pruebas de significación estadística de significación de la pendiente Sb rel (%) y los coeficientes de variación de los factores de respuesta.
Los límites de detección y cuantificación se determinaron calculando la pendiente de la curva de calibración (mAU vs mg/ml) así como la pendiente de la curva de desviación estándar (S) vs concentración y en ambos casos se extrapoló la respuesta a concentración cero y se obtuvo un estimado de la respuesta del blanco (Ybl) y de su desviación estándar (Sbl) respectivamente. Además se analizaron por triplicado muestras con cantidades cercanas a los límites calculados. Se determinó la media y la S.
RESULTADOS
Los resultados obtenidos en el estudio de especificidad del método, se ilustran en la Figura donde los cromatogramas muestran el pico correspondiente a la Mgf con un tiempo de retención de 9.2 minutos, así como los espectros UV del mismo. En los cromatogramas correspondientes al tratamiento de la muestra con medio oxidante, básico y ácido, se observa una disminución del pico correspondiente a la Mgf. los espectros UV de la Mgf en las muestras degradadas son similares a los del material de referencia químico.
Los resultados del estudio de precisión del método analítico muestran que en el estudio precisión intermedia, los valores de p resultaron ser mayores que 0.05, tanto para el test de Fisher como para la comparación entre las medias obtenidas los diferentes analistas y los diferentes días (Tabla 2). Por otra parte en el estudio de repetibilidad realizado, la media obtenida fue de 2,07 g /100g de hojas con un coeficiente de variación de 0,64 %.

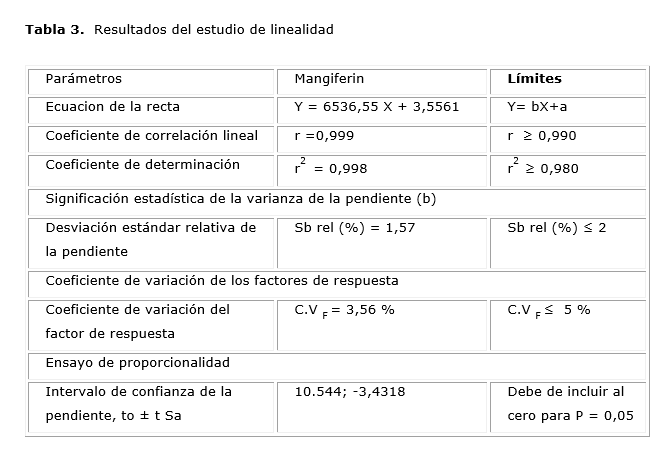
Los resultados de los estudios de linealidad se muestran en la Tabla 3.
Para el estudio de exactitud, la recuperación media fue de 99,41 %. Al compararse mediante una prueba de Student la recuperación media y 100 % de recuperación se obtuvo que la t experimental (texp= 1,50) es menor que la t tabulada (ttab= 2.306 para p= 0,05). Por otro lado, al aplicarse la prueba de la G de Cochran se obtuvo un valor de la G experimental (Gexp= 0,637) menor que la G tabulada (G tab= 0,797 para p= 0,05; K= 3 y n= 3).
En la Tabla 4 se muestran los Límites de detección (Ld) y de cuantificación (Lc) calculados. La recuperación promedio fue de 99,25 % con una Desviación Estándar de 0,23 %.
Es de resaltar que en todos los casos se obtuvo un coeficiente de variación menor que 2 %, los porcentajes de recuperación entre 98 y 102 %, que cumple lo establecido en la literatura.
DISCUSIÓN
El objetivo de un procedimiento analítico es determinar una información específica acerca de una muestra bajo investigación desde el punto de vista cualitativo y cuantitativo y esa información servirá para definir algunas características específicas de la fuente de origen de dicha muestra, permitiendo identificar parámetros de controles sobre la calidad de la misma.35
Diferente tipos de equipos e instrumentos pueden ser empleados en el proceso de análisis pero entre los más importantes que deben poseer los mismos, es que permitan definir una variable medible en correspondencia con los cambios que se produzcan en la muestras y por otro lado que pueda ser interpretada y controlada por el analista.35
Los métodos analíticos deben mostrar especificidad por la muestra, debe ser preciso y exacto. La repetibilidad de un método analítico está dado por la precisión en las medidas de las variables repetida bajo las mismas condiciones, mientras que la reproducibilidad está dado por la precisión en las medidas de las variables repetidas bajo diferentes condiciones.35
La cromatografía líquida de alta resolución es una herramienta para solucionar los inconvenientes de los métodos espectrofotométricos por su elevada sensibilidad y exactitud, además es en esencia un método separativo; lo que permite medir con gran selectividad el compuesto deseado, siempre y cuando se encuentre un sistema cromatográfico que asegure una adecuada separación.36,37 En el campo de la fitoquímica esto cobra especial importancia debido al gran número de compuestos con similitud estructural presentes en el material vegetal.
Los métodos que se establecen para la cuantificación de metabolitos en las diferentes fuentes deben ser confiables por lo que se someten a un proceso de validación que es la metodología establecida para la obtención de pruebas documentadas de que el método es lo suficientemente fiable para producir el resultado esperado.38,40
Los resultados obtenidos en el estudio de especificidad del método (Figura), demuestran la especificidad del método al no presentarse interferencias de picos adicionales en la determinación del metabolito de interés, no se evidencian interferencias de los productos de degradación.
En el caso de las muestras sometidas a diferentes condiciones de stress, se observa una disminución en el pico de la MgF, lo cual evidencia una degradación del compuesto. La similitud entre los espectros uv de los picos obtenidos para las muestras y el material de referencia, demuestra que estos se obtienen con elevada pureza, lo cual evidencia que no existe co-elución de metabolitos ni productos de degradación en el tiempo de retención de interés (9,2 minutos), por lo que nos encontramos ante un método selectivo.
En el estudio de precisión se demostró que en el caso de la repetibilidad los resultados analíticos cumplen con el límite establecido para el coeficiente de variación de métodos cromatográficos, el cual debe ser menor e igual que 2 %, esto demuestra que el método es repetible. Mientras que, al cumplirse los criterios de aceptación necesarios se comprueba que no existen diferencias significativas entre las dispersiones, ni entre las medias de ambos analistas, por lo tanto puede asumirse que existe homogeneidad en los datos obtenidos y de esta forma se demuestra que el método es preciso.
Los resultados de los estudios de linealidad muestran coeficientes de correlación y de determinación son superiores a los exigidos, 0,99 y 0,98 respectivamente, lo que demuestra con el valor del coeficiente de correlación obtenido, cercano a la unidad, la existencia de correlación con una probabilidad elevada, así como el grado de relación entre las variables concentración y respuesta detectada por el equipo empleado. También el coeficiente de variación de los factores de respuesta y la desviación estándar relativa de la pendiente fueron inferiores al normado como máximo para estos indicadores: 5 y 2 %, respectivamente; ambos son considerados estimadores puntuales que permiten caracterizar la variabilidad. El valor obtenido de los coeficientes de variación de los factores de respuesta, permite demostrar que existe variabilidad en la relación respuesta y concentración para cada nivel evaluado. El intervalo de confianza del intercepto incluye al cero, lo que permite excluir la significación del error del intercepto. Se demuestra con los resultados obtenidos la linealidad del método propuesto.
En el caso del estudio de la exactitud es de resaltar que en todos los casos se obtuvo un coeficiente de variación menor que 2 %, los porcentajes de recuperación entre 98 y 102 %, que cumple lo establecido en la literatura.39
Se comprobó además, que no existen diferencias significativas entre la recuperación media y 100 % de recobro, también que las varianzas de las concentraciones son equivalentes, se obtuvieron valores de G de Cochran experimental menor que la tabulada; por lo tanto puede asumirse que la concentración no influye en la variabilidad de los resultados.
El método fue suficientemente sensible, pues es capaz de cuantificar una cantidad equivalente al 87,5 % de degradación de la MgF por lo que el método podría ser utilizado en el estudio de conservación y estabilidad del material vegetal.
REFERENCIAS BIBLIOGRÁFICAS
1. Napralet Database, Univ. Ilinois, Chicago, IL, USA. [Citado 15 Febrero 2011] Disponible en URL: http://www.ag.uiuc.edu/ffh/narra.html
2. Desai PD, Ganguly AK, Govindachari TR, Joshi BS, Kamat VN, Manmade AH et al. Chemical investigation of some Indian plants: Part II. Indian Journal of Chemistry. 1966;4:457-549.
3. Garrido G, Gonzalez D, Delporte C, Backhouse N, Quintero G, Nunez-Selles AJ, et al. Analgesic and anti-inflammatory effects of Mangifera indica L. extract (Vimang). Phytother Res. 2001;15(1):18-21.
4. Makare N, Bodhankar S, Rangari V. Immunomodulatory activity of alcoholic extract of Mangifera indica L. in mice. J Ethnopharmacol. 2001;78(2-3):133-137.
5. Garcia D, Delgado R, Ubeira FM, Leiro J. Modulation of rat macrophage function by the Mangifera indica L. extracts Vimang and mangiferin. Int Immunopharmacol. 2002;2(6):797-806.
6. Garcia D, Leiro J, Delgado R, Sanmartin ML, Ubeira FM. Mangifera indica L. extract (Vimang) and mangiferin modulate mouse humoral immune responses. Phytother Res. 2003;17(10):1182-1187.
7. Martinez G, Delgado R, Perez G, Garrido G, Nunez Selles AJ, Leon OS. Evaluation of the in vitro antioxidant activity of Mangifera indica L. extract (Vimang). Phytother Res. 2000;14(6):424-427.
8. Sanchez GM, Rodríguez HMA, Giuliani A, Núñez Sellés AJ, Rodríguez NP, León Fernández OS, et al. Protective effect of Mangifera indica L. extract (Vimang) on the injury associated with hepatic ischaemia reperfusion. Phytother Res. 2003;17(3):197-201.
9. Sanchez GM, Re L, Giuliani A, Nunez-Selles AJ, Davison GP, Leon-Fernandez OS. Protective effects of Mangifera indica L. extract, mangiferin and selected antioxidants against TPA-induced biomolecules oxidation and peritoneal macrophage activation in mice. Pharmacol Res. 2000;42(6):565-573.
10. Sairam K, Hemalatha S, Kumar A, Srinivasan T, Ganesh J, Shankar M, et al. Evaluation of anti-diarrhoeal activity in seed extracts of Mangifera indica. J Ethnopharmacol. 2003;84(1):11-15.
11. Anila L, Vijayalakshmi NR. Flavonoids from Emblica officinalis and Mangifera indica - effectiveness for dyslipidemia. J Ethnopharmacol. 2002;79(1):81-87.
12. Aderibigbe AO, Emudianughe TS, Lawal BA. Antihyperglycaemic effect of Mangifera indica in rat. Phytother Res. 1999;13(6):504-507.
13. Aderibigbe AO, Emudianughe TS, Lawal BA. Evaluation of the antidiabetic action of Mangifera indica in mice. Phytother Res. 2001;15(5):456-458.
14. Rivera DG, Balmaseda IH, León AA, Hernández BC, Montiel LM, Garrido GG, et al. Anti-allergic properties of Mangifera indica L. extract (Vimang) and contribution of its glucosylxanthone mangiferin. J Pharm Pharmacol. 2006 Mar;58(3):385-92.
15. Tona L, Kambu K, Ngimbi N, Mesia K, Penge O, Lusakibanza M, et al. Antiamoebic and spasmolytic activities of extracts from some antidiarrhoeal traditional preparations used in Kinshasa, Congo. Phytomedicine. 2000;7(1):31-38.
16. El Ansari MA, Reddy KK, Sastry KNS, Nayudamma Y. Polyphenols of Mangifera indica. Phytochemistry. 1971;10(9):2239-2241.
17. El Ansari MA, Rajadurai S, Ayudamma Y. Studies on the polyphenols of Mangifera indica bark. Leather Science. 1967;14:247-251.
18. Bhatia VK, Ramanathan JD, Seshadri TR. Constitution of mangiferin. Tetrahedron. 1967;23(3):1363-1368.
19. Ramanathan JD, Seshadri TR. Constitution of mangiferin. Current Science. 1960;29(4):131-132.
20. Nigam SK, Mitra CR. Constituents of mango (Mangifera indica) roots. Indian Journal of Chemistry. 1964;2:378-379
21. Yoshimi N, Matsunaga K, Katayama M, Yamada Y, Kuno T, Qiao Z, et al. The inhibitory effects of mangiferin, a naturally occurring glucosylxanthone, in bowel carcinogenesis of male F344 rats. Cancer Lett. 2001;163(2):163-70.
22. Sato T, Kawamoto A, Tamura A, Tatsumi Y, Fujii T. Mechanism of antioxidant action of pueraria glycoside (PG)-1 (an isoflavonoid) and mangiferin (a xanthonoid). Chem Pharm Bull (Tokyo). 1992;40(3):721-4.
23. Ichiki H, Miura T, Kubo M, Ishihara E, Komatsu Y, Tanigawa K, et al. New antidiabetic compounds, mangiferin and its glucoside. Biol Pharm Bull. 1998;21(12):1389-90.
24. Guha S, Ghosal S, Chattopadhyay U. Antitumor, immunomodulatory and anti-HIV effect of mangiferin, a naturally occurring glucosylxanthone. Chemotherapy. 1996;42(6):443-51.
25. Yoosook C, Bunyapraphatsara N, Boonyakiat Y and Kantasuk C. Antiherpes simplex virus activities of crude water extracts of Thai medicinal plants. Phytomedicine. 2000;6(6):411-9.
26. Prabhu SN, Shyamala Devia CS. Efficacy of mangiferin on serum and heart tissue lipids in rats subjected to isoproterenol induced cardiotoxicity. Toxicol. 2006;228(2-3):135–139.
27. Toshihiro M, Hiroyuki I, Itsuko H, Naoki I, Motoshi K, Masayoshi K, et al. Antidiabetic activity of a xanthone compound, mangiferin. Phytomed. 2001;8(2):85–87.
28. Nong C, He W, Fleming D, Pan L, Huang H. Capillary electrophoresis analysis of mangiferin extracted from Mangifera indica L. bark and Mangifera persiciformis C. Y Wu et TL Ming leaves. J Chromatogr B. 2005;826(1-2):226–231.
29. Ting-Ting J, Maw-Rong L, Yi-Cheng C, Shu-Tuan C. Using LC/MS/MS to determine matrine, oxymatrine, ferulic acid, mangiferin, and glycyrrhizin in the Chinese medicinal preparations Shiau-feng-saan and Dang-guei-nian-tong-tang. J Pharm Biomed Anal. 2006;40(2):472-477.
30. Ronghua D, Kang L, Qing L, Kaishun B. Determination of Mangiferin, Jateorrhizine, Palmatine, Berberine, Cinnamic Acid, and Cinnamaldehyde in the Traditional Chinese Medicinal Preparation Zi-Shen Pill by High-Performance Liquid Chromatography. J Chromatogr Sci. 2004;42(4):207-210.
31. Geodakyan SV, Voskoboinikova IV, Kolesnik JA, Tjukavkina NA, Litvinenko VI, Glyzin VI. High-performance liquid chromatographic method for the determination of mangiferin, likviritin and dihydroquercetin in rat plasma and urine. J Chromatogr. 1992;577(2):371-375.
32. Rusakova SV, Kocherga SI. Photocolorimetric determination of mangiferin. Pharm Chem. 1977;11(5):723-724.
33. Krivut BA, Fedyunina NA, Kocherga SI, Rusakova SV. Spectrophotometric determination of mangiferin. Chem Natural Compounds. 1977;12:36-38.
34. Rybachenko AI, Krivut BA, Georgievskii VP. Fluorodensitometric determination of mangiferin and isomangiferin in Hedysarum flavescens and H. alpinum. Chem Natural Compounds. 1977;12:395-396.
35. Dierksneier G. Métodos cromatográficos. La Habana: Ed. Científico-Técnica; 2005.
36. Guidance for Industry. Analytical Procedures and Methods Validation. Chemistry, Manufacturing, and Controls Documentation. New York: FDA/Center For Drug Evaluation and Research; 2001.
37. International Conference on Harmonization. Validation of Analytical Procedures. Technical Requirements for the Registration of Pharmaceuticals For Human Use. Geneva: ICH-Q2A; 1995.
38. Farmacopea de los Estados Unidos. USP 30. The United States Pharmacopeial Convention. Versión electrónica. New York: Pharm Convention Inc.; 2007.
39. Castro M, Gascón S, Pujol M, Sans J, Vicente L. Validación de métodos analíticos. Madrid: Monografía AEFI. Hewlett Packard; 1989.
Recibido: 11 de julio de 2013.
Aprobado: 15 de febrero de 2014.
Lic. Jacqueline A. Romero Díaz. Centro de Investigación y Desarrollo de Medicamentos, Ave 26 # 1605, La Habana, Cuba.
Teléfonos: 8810882, 8811944, 8811424, Fax (537) 335556.
correo electrónico: aylema.romero@cidem.sld.cu

{kind=link}
{kind=link}