










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La eritrodermia fue descrita por primera vez en 1868 por Hebra, se caracteriza por eritema y escama en más del 90% de la superficie cutánea y es más común en hombres de 40 a 60 años. Se divide en primaria y secundaria; la primera es menos común, suele iniciar en el tronco y se generaliza en un lapso que va de días a semanas. La secundaria habitualmente se presenta por diseminación de una dermatosis previa; sin embargo, sus características usualmente se pierden, ya que la eritrodermia las enmascara.1
En esta dermatosis el reclutamiento masivo de células inflamatorias en la piel es de origen desconocido y el mecanismo fisiopatológico es diferente, dependiendo de la causa. También se han demostrado células inflamatorias en la circulación. Este infiltrado inflamatorio provoca un aumento en el recambio epidérmico con la consecuente dermatosis eritematoescamosa que provoca pérdida de proteínas, líquidos, electrolitos y ácidos nucleicos. La eritrodermia tiene múltiples etiologías y es un reto para el dermatólogo identificarlas. Las más comunes se engloban en el acrónimo PALM (psoriasis, dermatitis atópica, linfoproliferativa, la mayoría por linfoma que es el caso que presentamos y medicamentos).2,3
Los linfomas cutáneos primarios consisten en una proliferación anormal de linfocitos T o B que muestran tropismo por la piel, sin evidenciarse compromiso extracutáneo al momento del diagnóstico. Se dividen en linfomas de células T (75%-80%) y linfomas de células B (20%-25%). 4
El linfoma cutáneo primario de células T abarca a diferentes variantes de linfomas no-Hodgkin que se caracterizan por presentar linfocitos T atípicos con una presentación inicialmente cutánea y pronósticos diversos, dependiendo del subtipo y de la estadificación, su variante más frecuente, la micosis fungoide, constituye el 50% de todos los linfomas primarios de piel, su presentación clínica puede ser como máculas o placas de formas irregulares, tumores cutáneos o como eritrodermia exfoliativa clásica, tiene además múltiples variantes y un amplio diagnóstico diferencial, por lo que para su diagnóstico se requiere una estricta correlación entre la clínica y la histopatología. El síndrome de Sézary, por su parte, es considerado la variante leucémica de los linfomas cutáneos primarios y forma parte del diagnóstico diferencial de las eritrodermias, se caracteriza por la triada de eritrodermia, linfadenopatía y linfocitos atípicos circulantes con núcleo cerebriforme (células Sézary), es importante saber que en cualquier causa de eritrodermia se pueden encontrar células de Sézary sin que cumplan con los criterios para este síndrome; sin embargo> 20% de estas células circulantes sí constituyen parte de la triada diagnóstica y < 10% es un hallazgo inespecífico.4-6
En los linfomas cutáneos, la evaluación de ganglios linfáticos es una parte importante para el estadiaje, pronóstico y seguimiento. La afección ganglionar marca la evolución a enfermedad sistémica y la evidencia de adenopatía clínica determina la evolución de N0 a N1 en el caso del linfoma cutáneo de células T.2
A continuación, en este caso clínico mostraremos una de las variantes eritrodérmicas de los linfomas cutáneos, la micosis fungoide, que se convierte en un motivo para supresentación, que, por su variabilidad clínica, con frecuencia simula otras dermatosis.
Presentación del caso
Se presenta el caso de un paciente masculino de 72años de edad, procedencia rural, con antecedentes de hipertensión arterial para la cual lleva tratamiento con captopril y lepra lepromatosa multibacilar, ahora, en período de observación acude al servicio de dermatología, porque hace 1 año comenzó con enrojecimiento en toda la piel y le comenzaron a salir múltiples escamas en todo cuerpo con picazón intensa, por tal motivo se decide su ingreso para mejor estudio y tratamiento.
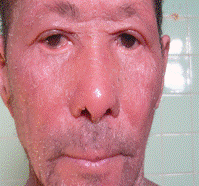
En el examen dermatológico presentaba lesiones universales eritemato escamosas, con eritema violáceo, escamas pardas, gruesas, secas y en su mayoría adheridas sobre todo en miembros inferiores y otras desprendibles, (Fig.1) además de exulceraciones postraumática en cara posterior de axila derecha con discreto exudado, como síntoma subjetivo prurito intenso generalizado.

A nivel de la cara, eversión de párpados inferiores (ectropión), con edema de los párpados de moderada intensidad y perdida de la cola de las cejas (Fig. 2).
En palma de manos se observa atrofia de la eminencia tenar e hipotenar (Fig. 3) y cierta tendencia a la reabsorción ósea de las falanges distales de algunos dedos. Además, trastornos de la sensibilidad mostrando hipoestesia a nivel de manos y pies.

El pelo se observaba ralo, opaco, quebradizo, con implantación y distribución según edad, sexo y raza y ausencia de bello a nivel corporal.
En las uñas las placas ungueales hiperquetosicas, distróficas en su porción distal, de color blanco amarillento en manos y pies.
Enel examen de cadenas ganglionares encontramos adenopatías axilares y regiones inguinales de aproximadamente 3 a4 cm, duras, firmes movibles, no dolorosas.
Ante la persistencia y características de las lesiones se decidió realizar estudios hemoquímicos, imaginológicos, biopsia cutánea y de ganglios linfáticos...
Laboratorio: HB: 10.5 g/l; Hto: 0.35;lec: 48 x 109/L, Pol 0. 20; L: 0.71; E: 0.1; M: 0.4; VSG: 15; Glu:4.2; Urea: 28; Crea: 61; Ac. Úrico: 357; TGO: 22; TGP:32.8; Col: 4.7,FA: 73; BT: 0.73; BD: 0.2; BI: 0.71; VDRL: no reactiva; Orina simple: sin alteraciones, Coagulograma con coagulo retráctil, plaquetas 215 x109; Proteínas Totales 55.6 mm/L, lo cual indica una hiperprotinemia ligera típica de la enfermedad , ante la anemia se realizó estudio de la misma, con Hb108 g/l, Hto0.35,CHCM 308, reticulocitos22 x109, prueba de Hook negativa, hipocromía x, plaquetas adecuada, lo que confirma una anemia por déficit de hierro por la perdida folatos. En los estudios imaginológicos, el ultrasonido de cadenas ganglionar con presencia de adenopatías inguinales bilaterales múltiples con una de mayor de tamaño. La radiografía de tórax el área cardiaca de límite máximo, aorta elongada, hilio engrosado de aspecto vascular que se corresponde con su cardiopatía hipertensiva, no lesiones pleuropulmonares senos costofrénicos libre.
Se realizaron biopsias de ganglio mostrando ganglio linfático inguinal linfadenitis dermatopatica (reticulosis hipomelanótica) y en el caso de lapiel reflejando linfoma no Hodgkin epidermotrófico focal compatible micosis fungoide. Otros complementarios por hematología especial como el medulograma con hipocromía dos cruces, anisopoiquilocitosis, leucocitosis con linfocitosis, plaquetas adecuada celularidad dos cruces, concluyendo como médula reactiva no concluyente para diagnóstico.
Durante el ingreso se vigilaron los signos vitales importantes en el paciente con eritrodermia, como tensión arterial, frecuencia cardíaca, frecuencia respiratoria y temperatura, además se indicó tratamiento por vía tópica con ácido salicílico y fomentos antiséptico en exulceraciones postraumática en cara posterior de axila derecha y por vía sistémica con esteroides y antihistamínicos.
Teniendo en cuenta las manifestaciones clínicas y los hallazgos histológicos se concluye el caso como una Micosis Fungoide, con seguimiento por Dermatólogos, así como internistas, hematólogos, oncólogos y médicos de atención primaria de salud.
Discusión
Los linfomas cutáneos de células T se consideran una neoplasia linfática crónica de la piel que puede afectar la sangre, los ganglios linfáticos y los órganos viscerales. Su diagnóstico preciso es importante, porque tiene similitud con otros infiltrados cutáneos linfocitarios y requiere un tratamiento específico para su control prolongado. Los linfomas de células T más comunes de la piel son la micosis fungoide (MF) que es el caso que presentamos dentro de su variabilidad clínica eritrodérmica y el síndrome de Sézary (SS). 2,7
La incidencia de la MF es 0,2 a 0,4 de casos nuevos por cada 100 000 habitantes, con predominio hombre-mujer de 2:1. Suele ser más frecuente en el cuarto o quinto decenio de la vida. La regla general es que se encuentra limitada a la piel durante muchos años. La causa es desconocida, pero han sugerido el papel de un antígeno crónico (posiblemente un retrovirus), estimulación de linfocitos T helper o CD4 por células de Langerhans intraepidérmicas en su génesis. Esta teoría sugiere que la causa de la enfermedad puede ser un proceso proinflamatorio o policlonal, hasta la bien desarrollada forma clonal de la enfermedad explicada por la persistencia inmunológica del microambiente cutáneo. Otra teoría ha sido en relación al porqué de la progresión de la micosis fungoide. Esta teoría trata de explicar que la enfermedad se inicia con una fase de exacerbación en la cual los linfocitos T de ayudas circulantes son reclutados en la dermis papilar. Posteriormente ocurre una selección de linfocitos T CD4 por citoquinas, la cual además promueve su proliferación hasta determinadas poblaciones clonales causantes de la neoplasia.8-11
La MF se caracteriza por presentar una progresión lenta y un curso prolongado de años a décadas generalmente indolente, con menos del 10% de los pacientes afectados evolucionando a etapas avanzadas de la enfermedad. El prurito es un síntoma prominente y el compromiso extra cutáneo, en caso de existir, generalmente solo se presenta en etapas tardías.
Se han descrito más de 30 variantes clínico patológicas de la MF, dentro de las más frecuentes se presentan la clásica, foliculotrópica, reticulosis pagetoide, piel laxa granulomatosa, hipopigmentada y eritrodérmica, esta última puede presentarse de novo o como progresión de las distintas etapas de la MF clásica, con eritema rojo brillante y descamación que compromete más del 80%-90%de la superficie corporal total, con prurito intenso y frecuentemente asociado a hiperqueratosis palmoplantar severa. A diferencia del síndrome de Sézary, no presenta adenopatías generalizadas ni células de Sézary circulantes. Otros síntomas y signos que pueden observarse en MF, particularmente en etapas avanzadas, son: compromiso del estado general, baja de peso, fiebre, insomnio (secundario a prurito), hiperqueratosis, descamación, fisuras en palmas y plantas, alopecia, ectropión. Cuando aparece compromiso extra cutáneo los órganos que con frecuencia están comprometidos son linfonodos, vísceras (particularmente pulmones, bazo e hígado) y sangre. La médula ósea usualmente no se afecta.2,12
El diagnóstico de este tipo de linfoma cutáneo de células T (MF) se hace por medio de una adecuada correlación clínico- histológica, utilizándose además otros métodos de laboratorio como la inmunohistoquímica que son una herramienta crucial, ya que permite establecer el inmunofenotipo, estudiando así los marcadores de superficie de los infiltrados linfociticos. Los linfocitos atípicos de la MF clásica se caracterizan por ser CD3+ (marcador de linfocito T), CD4+ (marcador de linfocitos T helper), CD8- y CD45RO+ (marcador de linfocitos T de memoria maduros). También puede presentar otros marcadores de células T para otras formas como (CD2, CD5 y CD7) que conforme avanza la enfermedad pueden ir perdiendo su expresión; particularmente la pérdida de CD7 se considera un hallazgo sensible y específico de MF. (13,14
El estudio de Etapificación de todo paciente con MF se considera esencial, en él se contemplan el examen físico completo: especial énfasis en el tipo de lesiones, porcentaje de superficie corporal total (SCT) comprometida y la búsqueda dirigida de adenopatías, organomegalias y masas. Otros estudios como Hemograma con determinación de células de Sézary, Citometría de flujo para células de Sézary: opcional en los casos con compromiso cutáneo menor al 10% de SCT; Análisis de reordenamiento del gen TCR en linfocitos desangre periférica: en caso de sospecha de compromiso sanguíneo; Panel metabólico; LDH; Tomografía axial computarizada (TAC) con contraste detórax, abdomen y pelvis o PET-CT: cuando hay compromiso cutáneo mayor al 10% de SCT, adenopatías palpables, exámenes de laboratorio anormales, transformación a células grandes o MF foliculotrópica. 6,15
Los objetivos del tratamiento de la MF son alcanzar y mantener la remisión, reducir la morbilidad y prevenir la progresión. Sin embargo, dado la naturaleza recurrente de la enfermedad, las respuestas suelen ser de corta duración, con recaídas frecuentes. Previo a indicar un tratamiento debemos considerar en primer lugar la etapa clínica de la enfermedad, ya que pacientes con etapas tempranas probablemente requerirán terapias tópicas, mientras aquellos en etapas más avanzadas se beneficiarán de tratamientos sistémicos. También es importante considerar determinados factores pronósticos específicos tales como la transformación a células grandes (requerirá terapias más agresivas) o el compromiso foliculotrópico (refractario a tratamientos tópicos por la profundidad y grosor del infiltrado). Además, hay que tener en cuenta la edad, comorbilidades, severidad de los síntomas y disponibilidad de terapias según recursos del centro y/o del paciente. (2,11,16
Las guías de la National Comprehensive Cancer Network (NCCN) clasifica las distintas opciones terapéuticas en 3 grandes grupos: terapias con efecto cutáneo directo: Corticoesteroides tópicos, Mostaza nitrogenada tópica (mecloretamina), Carmustina (BCNU), Retinoides tópicos, Fototerapia con el uso de 8-metoxipsoraleno oral + fototerapia UVA(PUVA) y Radioterapia, el segundo grupoque está basado en terapias sistémicas que se incluyen los Retinoides sistémicos, Interferones, Inhibidores de histonas de acetilasas como el vorinostat se administra vía oral mientras la romidepsina es de uso endovenoso, también este grupo los Antifolatos como el metotrexato por vía oral a dosis bajas; intramuscular o endovenoso a dosis altas y pralatrexato de forma endovenoso, los Quimioterapéuticos como gemcitabina, doxorrubicina liposomal, clorambucilo, pentostatina, etopósido, ciclofosfamida y temozolomida , los anticuerpo monoclonal humanizado contra CD52 que están presente en la superficie de células inmunes y Fotoforesis extracorpórea. El tercer grupo terapias combinadas muchas veces la combinación de terapias con efecto cutáneo directo con terapias sistémicas, o de 2 terapias sistémicas, es una estrategia útil en recaídas, casos refractarios, enfermedad extra cutánea o para potenciar las respuestas individuales de cada una. A su vez, no deben olvidarse las terapias de soporte tales como el manejo del prurito, control de infecciones, uso de emolientes, entre otros. 17,18

