










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introducción
El conocimiento de la estructura de los genes, cómo se regulan, transcriben y se traducen en las unidades funcionales de la célula, las proteínas, ha sido fundamental para empezar a comprender las causas y los mecanismos de producción de las enfermedades genéticas. Estas enfermedades constituyen un problema médico de primera magnitud, debido a que muchas de ellas causan graves trastornos a los pacientes y representan una carga física y emocional, y en algunos de los casos socioeconómica para las familias. Un grupo importante de estos factores afecta al sistema nervioso.1,2
La neurofibromatosis tipo I o enfermedad de Von Recklinghausen es una de las enfermedades genéticas que afecta el sistema nervioso, es la forma más frecuente dentro de los síndromes neurocutáneos denominados así por su origen común embrionario, es una enfermedad autosómica dominante, progresiva, de evolución impredecible, que afecta la piel y el sistema nervioso central y periférico; se debe a una mutación del cromosoma 17q11.2, codificante de una proteína supresora tumoral, la neurofibromina.3,4
La neurofibromatosis a pesar de ser una enfermedad sistémica tan frecuente y con capacidad para afectar a prácticamente la totalidad de las estructuras orgánicas, es poco conocida por los estudiantes de medicina y médicos generales, de ahí la importancia del conocimiento de estas. Para poder realizar un diagnóstico y tratamiento precoz así cómo brindar un adecuado asesoramiento genético a las familias afectadas, consideramos de utilidad realizar la presentación de un paciente de 25 años de edad, con el diagnóstico de neurofibromatosis tipo I.
Presentación del caso
Paciente masculino de 25 años de edad, de procedencia urbana, diestro, hijo único de padres no consanguíneos, el cual comenzó a notar presencia de tumoraciones subcutáneas en los cuatro miembros y en la región dorsovertebral, no dolorosas, blandas y movibles.
Poco después, presentó de forma progresiva, parestesias y dolor en la región dorsolumbar y en ambos miembros inferiores, así como aumento de volumen a nivel de todo el miembro inferior derecho, lo que le dificulta la marcha.
Por esta sintomatología ingresó en el Hospital General Docente “Dr. Agostinho Neto” donde se le realizaron estudios radiológicos detectándose un paquete tumoral en el área paravertebral y región inguinal; se decidió realizar tratamiento quirúrgico para extracción y biopsia de las lesiones, informándose presencia de neurofibromas.
Examen físico neurológico (datos positivos)
Marcha antiálgica, atrofia muscular supra e infraclavicular, supra e infraespinosa, interósea, tenar e hipotenar bilateral. Tumoraciones subcutáneas múltiples, a nivel del cuello, extremidades y tronco (Figura 1).

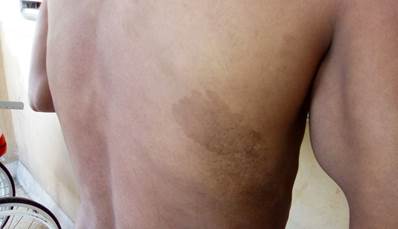
Se encontraron, además, varias manchas color “café con leche” multiformes, con diámetro oscilando entre los 7 mm y hasta los 24 mm (Figura 2).
Disminución de la fuerza muscular 4/5 próximo distal de las cuatro extremidades, así como atrofia interósea bilateral (Figura 3).

Hiperreflexia generalizada de los reflejos osteotendinosos. Examen biomicroscópico en Lámpara de Hendidura: ambos ojos: se aprecian múltiples nódulos de Lisch de aproximadamente 1 mm, redondeados, color carmelita en el estroma iridiano (Figura 4).

Fig. 4 Nódulos de Lisch de aproximadamente 1 mm, redondeados, color carmelita en el estroma iridiano.
Resonancia magnética nuclear dorsolumbar T2 vista lateral se observan múltiples neurofibromas, (Figura 5).

Discusión del caso
Las neurofibromatosis constituyen un grupo de enfermedades neurocutáneas que muestran extrema heterogeneidad clínica y están caracterizadas por anormales crecimientos en tejidos derivados de la cresta neural embriogénica.5 Las dos formas clínicas principales que existen son la tipo 1 (NF1) o enfermedad de von Recklinghausen, causada por un defecto en el brazo largo del cromosoma 17 (17q 11.2) y la tipo 2 (NF2) donde las mutaciones ocurren en el cromosoma 22 (22q 12).6
En la tipo 1 ocurre la inactivación del neurofibromin. El mapa genético del NF1 pertenece al cromosoma 17, su RNA contiene una proteína llamada neurofibromin, que al ser un GTP activador puede controlar un inhibidor de la p21-ras protooncogen, el cual desempeña una función central en las vías señaladas como mitogénicas.7
Se cree que el gen de la neurofibromatosis tipo 1 produce una proteína similar a las proteínas activadoras del Ras-GTPasa, que a su vez están involucradas en los mecanismos que controlan el crecimiento y la diferenciación celular mediante la interacción con la familia del gen Ras. Esta forma mutante sirve como catalizador para la formación tumoral a través de señales de transducción alteradas.8
La incidencia es de 1/3,000 recién nacidos y la prevalencia se sitúa en torno a 1/50,000 habitantes, presentando una elevada proporción de nuevas mutaciones, aproximadamente el 50 %. La penetrancia es del 100 % con una expresividad variable.9
Los neurofibromas presentes en la NF-1 son de tres tipos bien diferenciados tanto clínica como histológicamente:
Neurofibromas intradérmicos benignos, presentes en el 95 % de los pacientes.
Neurofibromas nodulares en los nervios periféricos de cualquier localización que, aunque no infiltran los tejidos circundantes, pueden crecer enormemente y provocar síntomas por compresión.
Neurofibromas plexiformes, que habitualmente son congénitos y están presentes en el 30 % de los pacientes con NF-1.
Estos tumores representan la mayor causa de morbilidad y mortalidad, ya que afectan a extensas porciones de los nervios, infiltrando tanto al nervio como a los tejidos circundantes. Todo ello conlleva una desfiguración general y complicaciones de tipo mecánico. Alrededor del 2-16 % de los pacientes que presentan neurofibromas nodulares y plexiformes sufren un empeoramiento ocasionado por la transformación en tumores malignos de las vainas de los nervios periféricos.10
Lo más común es que el diagnóstico se haga durante la niñez, por la existencia de antecedentes familiares de este trastorno, aunque como observamos en nuestro caso no se recogen antecedentes familiares, por lo que estamos en presencia de una mutación de novo y además por las manifestaciones cutáneas obvias, como las manchas de color marrón claro y neurofibromas. Por lo general, las lesiones cutáneas, son asintomáticas y no representan peligro. Los hamartomas de iris (nódulos de Lisch) son difíciles de constatar en los primeros años y son asintomáticos11; dado que no se ven en personas sanas, se consideran patognomónicos de NF1 y lo pudimos constatar en nuestro paciente.
Entre las alteraciones esqueléticas destaca displasia de alas del esfenoides, arqueamiento congénito o pseudoartrosis, quistes óseos, hipercrecimiento óseo y la cifoescoliosis. (12
El diagnóstico de la NF-1 se basa en los criterios clínicos establecidos por el National Institute of Health Consensus Development Conference en 198813 y se establece el diagnóstico con dos o más de estos signos.
Criterios diagnósticos de la neurofibromatosis tipo I:
Seis o más manchas color “café con leche” de 1,5 cm o mayores en pospúberes y de 0,5 o mayores en prepúberes.
Dos o más neurofibromas de cualquier tipo o uno o más neurofibromas plexiformes.
Pecas en las axilas y/o en las ingles.
Gliomas de las vías ópticas.
Dos o más nódulos de Lisch (hamartomas benignos del iris).
Una lesión ósea distintiva (displasia del esfenoides o displasia o adelgazamiento de la cortical de hueso largo).
Un familiar de primer grado con NF1.
Dado el avance de las técnicas diagnósticas, a los criterios diagnósticos debería añadirse un octavo criterio: la presencia de lesiones hiperintensas en la resonancia magnética potenciada en T2, que se encuentran en el 60-70 % de los niños con NF1.
En el caso en estudio no aparecieron lesiones encefálicas intraparenquimatosas pero si se encontraron lesiones muy notables en la columna dorsolumbar a nivel de los agujeros de conjunción, lo que explicaba las manifestaciones clínicas del paciente, tales como, la atrofia, la impotencia funcional en ambos miembros inferiores y la deformidad esquelética.
El tratamiento de la neurofibromatosis tipo 1 debe ser individualizado; depende de la localización del tumor, si causa dolor o desfiguración importante, si afecta la función y de su velocidad de crecimiento. Las opciones van desde la observación, la resección quirúrgica parcial o total y el uso de quimioterapia.
Consideraciones finales
Dada la alta incidencia de esta enfermedad dentro de los trastornos hereditarios, de ser una enfermedad sistémica y progresiva, aunado al escaso conocimiento teórico del personal médico en la sospecha diagnóstico y del costo que representan los estudios de neuroimagen, es necesario enfatizar en las manifestaciones clínicas para que pueda ser diagnosticada tempranamente, lo que favorecerá la calidad de vida de estos pacientes así como la posibilidad de brindar un adecuado asesoramiento genético.

