










My SciELO
 Custom services
Custom servicesServices on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMEDISAN
On-line version ISSN 1029-3019
MEDISAN vol.20 no.3 Santiago de Cuba Mar.-Mar. 2016
TESTIMONIO
Un niño extraordinario fallecido a causa del síndrome de Hurler-Scheie
An extraordinary deceased child due to the Hurler-Scheie syndrome
Lic. Yannelis Martínez Yero I y Dra. Dulce María Echavarría Estenoz II
I Facultad No. 1, Universidad de Ciencias Médicas, Santiago de Cuba, Cuba.
II Hospital Docente Infantil Sur, Universidad de Ciencias Médicas, Santiago de Cuba, Cuba.
RESUMEN
Se expone la experiencia personal de una madre que debió enfrentar el síndrome de Hurler-Scheie, rara enfermedad padecida por su hijo ya fallecido, con vistas a demostrar que la observación de los cambios producidos en un niño, es vital para su desarrollo. El cuidado hacia un menor con esta afección resulta indispensable para lograr una infancia con calidad de vida; por tanto, el sistema de salud cubano debe continuar diseñando estrategias que garanticen un diagnóstico precoz y la posibilidad de alternativas terapéuticas oportunas como la terapia de reemplazo enzimático.
Palabras clave: mucopolisacaridosis, deficiencia enzimática, enfermedad metabólica congénita, glicosaminoglicano, hepatomegalia, apnea del sueño, rigidez articular, terapia de reemplazo enzimático.
ABSTRACT
The personal experience of a mother that faced the Hurler-Scheie syndrome, strange disease suffered from his son already deceased, is exposed with the purpose to demonstrate that the observation of the changes occurring in a child, is vital for his development. The care toward a young child with this disorder is indispensable to achieve a childhood with life quality; therefore, the Cuban health system should continue designing strategies that guarantee an early diagnosis and the possibility of opportune therapeutic alternatives as the enzymatic substitution therapy.
Key words: mucopolysaccharidosis, enzymatic failure, congenital metabolic disease, glycosaminoglycan, hepatomegaly, sleeping apnea, joint stiffness, enzymatic substitution therapy.
INTRODUCCIÓN
En este trabajo se expone la experiencia personal de una madre que debió enfrentar la rara enfermedad que padeció su hijo Fabio Alejandro Barthelemy Martínez, ya fallecido, conocida como síndrome de Hurler-Scheie (una MPS I), a fin de motivar a futuros padres para que se informen sobre el tema, pues aún sin tener un antecedente genético familiar se corre el riesgo de tener un niño con esas características; incentivar a los especialistas de la salud a profundizar en el conocimiento de los síntomas que dicha afección puede producir desde sus inicios; demostrar que una adecuada observación de los cambios que puede presentar un niño, sobre todo en su primera etapa de desarrollo, es vital; mostrar que la búsqueda temprana de ayuda profesional puede marcar la diferencia para lograr un tratamiento oportuno que mejore la calidad de vida del infante, así como reflexionar sobre lo importante que resulta prevenir esta enfermedad a través de los métodos de diagnóstico prenatal y preconcepcional existentes.
PRIMER AÑO DE VIDA
Mi primer bebé fue un niño muy esperado, él llenaba con alegría y amor cada minuto de las vidas de las personas que constituían mi núcleo familiar: padre, madre y hermano.
Al analizar los antecedentes patológicos familiares se halló que no existían enfermedades hereditarias, defectos congénitos, consanguinidad, ni manifestaciones de retraso mental.
Entre los antecedentes prenatales figuraron:
- Embarazo sano y sin riesgo
- Asistí a todas las consultas médicas y me realizaron todos los exámenes complementarios en los diferentes periodos del embarazo, cuyos resultados fueron normales; por esa razón me consideraron embarazada con riesgo genético bajo.
- Durante toda esa etapa no tuve ninguna infección o enfermedad, anemia, fiebre, no sufrí ninguna agresión del medio, ni tomé medicamentos.
Antecedentes perinatales:
- El parto se me presentó a las 39,3 semanas y para evitar el sufrimiento del feto, pues no pude dar a luz de forma natural, se decidió realizar cesárea.
- El niño tuvo un llanto fuerte al nacer.
- Presentó todas las mensuraciones dentro de los límites normales con una talla de 50 cm y un peso de 3340 g.
- Tuvo una succión vigorosa.
- Orinaba y defecaba sin problemas.
Nació un niño normal; por tanto, no nos imaginamos lo que nos depararía el destino. Al parecer sería muy inteligente, pues desde muy temprano comenzó a emitir sus primeros sonidos e intentaba hablar. Fue siempre muy comunicativo y estaba pendiente de todo lo que ocurría a su alrededor.
Pensamos que todos los problemas de salud presentados durante su primer año de vida eran propios de la edad y no le dimos la mayor importancia, pero buscamos ayuda profesional y cumplíamos cada tratamiento a cabalidad. Comenzó a mostrar problemas respiratorios como catarros con frecuencia siempre acompañados de una ligera falta de aire, motivo por el cual tuvo algunos ingresos y cuando no fue hospitalizado se recuperaba mediante un tratamiento médico que cumplíamos al pie de la letra.
A medida que pasaba el tiempo continuaron los cambios en su crecimiento, entre ellos: episodios de diarreas a la edad de 7 meses, por lo cual sufrió una leve deshidratación y requirió ser hospitalizado, pero la rebasó rápidamente. Comenzó a presentar debilidad en los miembros inferiores y superiores de manera general. Gateaba con dificultad, a tal punto, que nunca pudo hacerlo bien, pues sus brazos no podían sostener su cuerpo y siempre se caía hacia delante. Cuando lo cargaba, su espalda tendía a encorvarse, parecía no tener fuerzas para mantenerse erguido como otros niños, por lo que en la parte media se le comenzó a desarrollar una pequeña curvatura. No podía sentarse por sí solo cuando tenía la edad de 9 meses, puesto que se caía hacia los lados sin fuerzas suficientes como para mantenerse sentado.
Tras varios intentos por buscar la causa de sus ligeras dificultades, los médicos descartaron la probabilidad de cualquier enfermedad e indicaron un tratamiento de rehabilitación con fisioterapia, que pensamos corregiría esos problemas físicos, pues nunca se pensó en una enfermedad tan grave.
A partir de los 10 meses comenzó a dar indicios de un poco más de fortaleza y ya lograba sentarse con ayuda, aunque en ocasiones lo hacia por sí solo. En ese mismo tiempo, comenzaron sus primeros intentos para ponerse de pie, para lo cual se ayudaba de los objetos que tenía a su alrededor. Continuó su rehabilitación y pasados unos meses, a la edad de un año, daba ya sus primeros pasos con ayuda. Siempre necesitó de nuestro apoyo para caminar, sentarse y pararse, estas 2 últimas acciones fueron muy difíciles para él y no llegó a caminar con independencia hasta la edad de 1 año y 5 meses de nacido (figura 1).

Parecía un niño sano, era hermoso, alto para su edad, llamaba mucho la atención de todo el que lo conocía, pues su crecimiento fue muy rápido en comparación con otros niños y creíamos que el proceso por el cual atravesaba en ese momento se resolvería con su rehabilitación en fisioterapia, que fue de gran ayuda en esta etapa de su vida. Entonces continuamos muy esperanzados de que mejoraría totalmente y así seguimos hasta que cumplió los 2 años de edad.
DESCUBRIMIENTO DEL SÍNDROME DE HURLER-SCHEIE
Al llegar a la edad de 2 años, en mi bebé continuaron los cambios físicos que lejos de mejorar, lo empeoraron. Su fisonomía comenzó a cambiar un poco, era evidente que algo andaba mal y notamos lo siguiente:
- Sus rasgos faciales comenzaron a tornarse cada vez más gruesos y toscos con un puente nasal bajo.
- Las córneas se volvieron opacas.
- Su cabeza comenzó a ser un poco más grande que el promedio para su edad.
- Su cuello era muy corto.
- Con el tiempo su crecimiento fue interrumpido poco a poco.
- La rigidez se hizo evidente en todas las articulaciones: manos, muñecas, hombros, codos, caderas, rodillas, tobillos y el movimiento comenzó a limitarse bastante.
- Se paraba y caminaba un poco encorvado debido a la rigidez de las articulaciones de las caderas, hombros, rodillas y la marcha era dificultosa.
- Sus manos eran pequeñas, anchas, con dedos gruesos que gradualmente se curvaron un poco.
- Periódicamente tenía diarreas y heces blandas.
Todos estos cambios nos alertaron de que era necesario un chequeo médico más profundo.
Repentinamente, a los 2 años, el niño tuvo otro episodio de diarreas, por lo cual fue hospitalizado, sin encontrar una causa aparente. Cuando le dieron el alta me dirigí al Centro de Genética Médica Provincial de Santiago de Cuba, donde fui atendida rápidamente y para mi asombro se me comunicó que la impresión diagnóstica en ese momento era de una mucopolisacaridosis, específicamente el síndrome de Hurler (figura 2).

Comenzaron así los primeros análisis para corroborar aquella impresión diagnóstica. A petición personal fue remitido al Centro de Genética Nacional en Ciudad de la Habana, donde después de varios análisis a través del tiempo, se logró realizar la determinación enzimática, lo cual confirmó que el niño tenía el síndrome de Hurler-Scheie (MPS I).
Debido a que esta enfermedad es muy rara y poco conocida en Cuba fue muy difícil saber qué hacer al comienzo. Como madre me di a la tarea de estudiarla profundamente, entonces comencé a buscar y experimentar alternativas que ayudaran a mejorar su calidad de vida. Pude aprender que esta afección genética es de difícil diagnóstico, puesto que los niños nacen aparentemente sanos y los síntomas comienzan a aparecer con más fuerza a la edad de 2 o 3 años, según la gravedad.
Aprendí además, que el síndrome de Hurler-Scheie pertenece al grupo de enfermedades conocidas también como mucopolisacaridosis (MPS) o enfermedades lisosomales por acumulación. Este es el nombre dado a un grupo de afecciones raras hereditarias, causadas por la ausencia o el mal funcionamiento de ciertas enzimas necesarias para el procesamiento o degradación de largas cadenas de moléculas de azúcar, también conocido como glicosaminoglicanos (GAGs) en un ciclo continuo de producción y reciclaje necesario para mantener la salud, lo cual significa que se transmite de padres a hijos. Ambos progenitores necesitan transmitir el gen defectuoso para que su hijo desarrolle este síndrome.1
Varios autores1,2 refieren que esta afección es un trastorno metabólico o sea una enfermedad metabólica congénita. Un desorden de carácter autosómico recesivo que resulta de mutaciones en el gen codificador de la enzima alpha L-iduronidasa, por lo cual se produce una acumulación de los GAGs heparán sulfato y dermatán sulfato en todos los tejidos del organismo y causan una amplia variedad de síntomas físicos, así como anormalidades que dañan los órganos, entre ellos el corazón. Estas moléculas de mucopolisacáridos se encuentran en todo el cuerpo, a menudo en las secreciones mucosas y en el líquido que rodea las articulaciones. La tasa de acumulación es variable en cada persona afectada y esto hace que haya una gran diversidad en las manifestaciones clínicas, cuyos síntomas se hacen más aparentes a medida que avanza dicha acumulación.
ENFRENTAMIENTO A LA ENFERMEDAD
El niño comenzó a tener diferentes problemas de salud, pero de manera general era fuerte y en esta etapa no tuvo muchas hospitalizaciones. A pesar de tener episodios de catarro y falta de aire en varias ocasiones, su vida resultó bastante tranquila.
Uno de los primeros problemas que comenzó a experimentar y le ocasionó las mayores limitaciones fue la rigidez que afectó cada una de sus articulaciones, debido a:
- La limitación de amplitud articular en codos y hombros no le permitía rascar bien su cabeza ni otras partes del cuerpo a las cuales no alcanzaba, por ello dependía casi siempre de mi colaboración para poder acceder a ellas.
- Le resultaba imposible hacer un puño con los dedos de sus manos, tenía muy poca presión al agarrar los objetos, pero aún así se esforzaba para hacerlo.
- Con mucha dificultad se ponía o quitaba cualquier prenda de vestir, pues su limitación se lo impedía.
- Tenía displasia de cadera.
- No podía amarrar sus zapatos y le era casi imposible abotonar una camisa.
- Tenía un genus valgo, que unido a la rigidez articular de sus rodillas, provocó que sus piernas siempre estuvieran en flexo al estar de pie, de manera que llegaron a alcanzar un ángulo aproximado de 45 grados y esto le provocaba dolores en esas partes de su cuerpo si permanecía de pie, aún por un corto periodo de tiempo.
Teniendo en cuenta esos problemas de salud y para ayudarlo a mejorar su condición física, se le indicó un nuevo tratamiento con fisioterapia, la cual contribuyó, de cierta forma, a mejorar cada uno de sus movimientos, así como su resistencia física durante el juego y la marcha (figura 3).
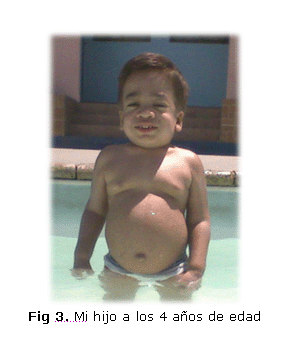
A pesar de ello, estas afectaciones limitaron su desarrollo en actividades de la vida cotidiana donde siempre buscaba apoyo mientras jugaba o caminaba. A medida que pasaba el tiempo su limitación iba en aumento y su resistencia física al caminar o cuando intentaba correr era cada vez menor, apenas soportaba 5 minutos de marcha, pues experimentaba cierta fatiga. Desde los 2 años hasta el último momento de su vida, su sistema respiratorio se afectó cada vez más, evidenciado no solo en su respiración ruidosa sino en los instantes de apnea del sueño que experimentaba al dormir, parecía como si dejara de respirar.
Muchas veces tuvo infección respiratoria y le resultaba muy difícil expulsar las secreciones, debido a que eran muy viscosas. Para lograr una respiración más estable durante el sueño elevábamos la cabecera de su cama a casi 45 grados. También experimentaba cianosis alrededor de sus labios y en la punta de sus dedos, provocada por la apnea del sueño antes referida, fundamentalmente durante los procesos de infecciones respiratorias, pues quedaba sin respirar por varios segundos y luego regresaba a la normalidad.
Este problema respiratorio desafortunadamente afectó también su corazón y, según el diagnóstico de los especialistas, desarrolló una miocardiopatía hipertrófica no obstructiva. Su hepatomegalia contribuyó a que tuviera un abdomen globuloso. Su visión parecía ser limitada en la noche, manifestaba no ver bien en ambientes con cierta oscuridad, e incluso, en este horario se le dificultaba reconocer a determinada distancia, alguna persona conocida.
Una buena higiene era crucial; su sudoración era tan abundante que parecía fuera de lo común, esto también sucedía en una habitación climatizada, donde la ropa que llevaba puesta parecía como si estuviera húmeda. Durante el sueño sus sábanas quedaban bañadas por el sudor. De ahí que tratáramos de mantenerlo con ropa fresca y en un ambiente con la mayor ventilación posible.
Recibió una alimentación muy sana sin limitaciones, con aquellos alimentos que aportaran vitaminas, minerales y todos los nutrientes necesarios para una dieta balanceada; nunca detectamos que algo le hiciera daño. Todo esto lo ayudó a mantenerse sano en otros aspectos y a ganar peso adecuadamente, aunque muchas veces aumentaba fácilmente y superaba el peso adecuado para su edad. Logramos ayudarlo a erradicar sus episodios de diarrea con la dieta referida anteriormente y la incorporación del yogur natural. Esa combinación le hizo evacuar sus necesidades fisiológicas de forma normal. Para poder ingerir los alimentos había que darle pequeñitas porciones, pues manifestaba que no podía tragar lo que representaba una cucharada, sentía como si se ahogara, por eso era muy cuidadoso y no permitía que le dieran grandes porciones a la vez.
Cada día era una batalla a librar por su enfermedad y la experiencia diaria me fue dando un poco de información vital para mejorar su calidad de vida.
VIVIR SIN LIMITACIONES
Aunque enfrentar esta enfermedad era algo muy difícil, siempre le enseñamos a tener una autoestima muy elevada, nunca le hicimos saber que sus dificultades físicas limitaban una vida normal y feliz.
La característica más significativa de su persona y por la que todos lo conocieron resultó ser su inexplicable inteligencia; tuvo un coeficiente superior al de la edad por la cual transitaba. Desde pequeño su lenguaje fue muy claro y cada vez incorporaba a su vocabulario una amplia gama de palabras, que en su mayoría no eran propias de un niño de su edad.
Se inclinó mucho al uso de la tecnología; sabía utilizar el celular para múltiples funciones y aprendió a trabajar en la computadora con juegos y programas educativos dedicados a niños de su edad; también aprendió a manipular otros equipos electrodomésticos con destreza. Él podía entender lo que veía y ciertamente tenía una memoria exquisita; era un niño muy precoz y observador, capaz de llegar a sus propias conclusiones.
Nunca fue excluido de la sociedad. Uno de los escenarios de actuación más importantes de su vida fue la escuela, donde demostró su inteligencia y participó junto a sus compañeritos en todas las actividades escolares y extraescolares desarrolladas. Tuvo muy buena relación con sus maestras y logró ser aceptado en un colectivo de niños, que lo cuidaron, entendieron y apoyaron; allí también aprendió a compartir y a vivir en sociedad.
UNA LECCIÓN DE VIDA
Es muy difícil para una madre saber que la vida de un hijo se le va entre sus manos sin poder decidir su curso. En esta oportunidad fue el mío, quien sufrió y murió a causa de esta devastadora enfermedad (figura 4), pero mañana pueden nacer otros infantes con problemas similares y todos tienen el derecho de recibir un tratamiento que les ayude a mejorar su vida.

Tristemente esta afección le arrebató la vida el 25 de mayo del 2014, a la edad de 5 años, justo un mes antes de cumplir los 6. Murió producto de una complicación respiratoria, una bronconeumonía, la cual propició que fallara su corazón y le ocasionó una arritmia que conllevó a un paro cardiaco, sin poder rebasar esta gravedad. Lo más doloroso de todo este proceso fue el diagnóstico tardío de la enfermedad y la ausencia de un tratamiento para combatirla.
De hecho, enfrentar este problema me permitió compartir el dolor de las madres que pudieran estar en mi situación. Cada padre debe convertirse en el propio médico de su hijo, ser capaz de autoprepararse e interpretar cada cambio que experimente; por esa razón, el empeño que padres y médicos tengan para descubrir cualquier anomalía en un bebé, sin lugar a dudas será crucial en una detección temprana de la enfermedad, lo cual es vital para obtener mejores resultados durante su tratamiento.
El asesoramiento genético ayuda a los padres con antecedentes familiares de mucopolisacaridosis a determinar si son portadores de un gen mutado que causa dichos trastornos. Es por ello, que tanto los especialistas en esta rama como los pediatras, deben profundizar en todo lo relacionado con la enfermedad para brindar una orientación adecuada a los progenitores, de manera que estos puedan enfrentar el problema.
Cuando un niño tiene esta afección genética debe ser atendido por un equipo multidisciplinario de médicos especialistas que trabajarán juntos para elaborar un plan terapéutico. Resulta importante incluir al genetista, cuyos consejos y apoyo serán útiles tanto para el niño como para la familia.
Desde mi modesto punto de vista, en este sistema de salud deben diseñarse estrategias en aras de garantizar una detección temprana de esta enfermedad, a través de los métodos concepcionales y preconcepcionales existentes y lograr así un diagnóstico precoz, que es de vital importancia para la aplicación de un tratamiento que contribuya a mejorar la calidad de vida de cada niño afectado.
A pesar de ser una afección muy rara no deja de existir, es por ello que llamo a la lucha para unir esfuerzos, que garanticen la posibilidad de alternativas de tratamiento, entre los cuales se encuentra la terapia de reemplazo enzimático, a fin de que estos niños sean beneficiados y no tengan limitaciones en este sentido. Dicha terapia puede ayudar a que cada infante disfrute de una vida normal.
En el 2003, la Agencia de Drogas y Alimentos (FDA, por sus siglas en inglés) aprobó el uso de la terapia de reemplazo enzimático (TRE) con iduronidasa, obtenida por ingeniería genética.
- Es un tratamiento específico que, mediante la administración suficiente de la enzima deficitaria, trata de evitar o revertir la acumulación de glicosaminoglicanos intracelulares. Consiste en la administración semanal de iduronidasa recombinante humana (laronidasa) en una dosis de 100 unidades/kg/semana por vía endovenosa (0,58 mg/kg/semana).
- Hasta la fecha, se ha demostrado que es un tratamiento seguro y efectivo.
- Los beneficios comprobados incluyen mejoría de la función pulmonar con aumento de la capacidad vital y la distancia caminada en una prueba de 6 minutos en las formas moderadas y leves (Hurler-Scheie y Scheie).
- Disminuye significativamente la hepatoesplenomegalia.
- Aumenta la movilidad de hombros y codos.
- Disminuye el índice de apneas e hipopneas durante el sueño.
- Disminuye la eliminación de glicosaminoglicanos urinarios y otros.2-4
Desde 1980 se han realizado a escala mundial más de 300 trasplantes de médula ósea (TMO) alogénicos en pacientes con MPS I. Este trasplante corrige la deficiencia enzimática en los afectados, pues reemplaza el sistema monocitomacrófago deficiente por otro (del donante), capaz de secretar la enzima. Previene la progresión de la enfermedad al disminuir la acumulación de GAG en los tejidos. Se ha utilizado en algunos pacientes con la citada afección y el tratamiento ha tenido diferentes resultados.2-4
Desafortunadamente mi hijo no pudo recibir un tratamiento adecuado que lo ayudara a tener una mejor calidad de vida y aunque en el mundo existen alternativas, como la terapia de reemplazo enzimático, el sistema de salud cubano no cuenta con los recursos necesarios para este tipo de terapia, debido a que es muy cara y difícil de adquirir.
Toda la experiencia vivida ciertamente constituye una lección de vida, pues mi niño fue un ejemplo de fortaleza, perseverancia, inteligencia, optimismo, pero lo más importante fue el amor a su familia, amiguitos y a la vida. Él me enseñó a no darme por vencida jamás y luchar ante toda circunstancia por lo más preciado que tiene el ser humano, "la vida".
REFERENCIAS BIBLIOGRÁFICAS
1. Grupo de trabajo de enfermedades poco frecuentes. Consenso de diagnóstico y tratamiento de la mucopolisacaridosis de tipo I. Arch Argent Pediatr. 2008; 106(4):361-8.
2. Miebach E. Enzyme replacement therapy in mucopolisaccharidosis type I. Acta Paediatr Suppl. 2005; 94(447):58-60.
3. Wraith JE, Beck M, Lane R, van der Ploeg A, Shapiro E, Xue Y, et al. Enzyme replacement therapy in patients who have mucopolysaccharidosis I and are younger than 5 years: results of a multinational study of recombinant human alfa-L-iduronidase (Laronidase). Pediatrics. 2007; 120(1):e37-46.
4. Muenzer J, Fisher A. Advances in the treatment of mucopolysaccharidosis Type I. N Engl J Med. 2004; 350(19):1932-4.
Recibido: 17 de diciembre de 2015.
Aprobado: 7 de enero de 2016.
Yannelis Martínez Yero. Facultad No. 1, Universidad de Ciencias Médicas, avenida de las Américas, entre calles I y E, reparto Sueño, Santiago de Cuba, Cuba. Correo electrónico: yannelis.martinez@sierra.scu.sld.cu

