










My SciELO
 Custom services
Custom servicesServices on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkMEDISAN
On-line version ISSN 1029-3019
MEDISAN vol.21 no.11 Santiago de Cuba Nov. 2017
CASO CLÍNICO
Acardius mylacephalus: un diagnóstico poco frecuente
Acardius mylacephalus: an infrequent diagnosis
Dra. Valia Hernández Viel, Dr. Alexander Llauger La Rosa y Dra. Margarita Argüelles Arza
Centro Provincial de Genética, Hospital Infantil Docente Sur "Dr. Antonio María Béguez César", Santiago de Cuba, Cuba.
RESUMEN
Se presenta el caso clínico de una primigesta de 21 años de edad, a quien se le detectó un embarazo gemelar monoamniótico y monocigótico en el examen ecográfico en el Centro Provincial de Genética de Santiago de Cuba. El primer feto presentaba hidronefrosis bilateral predominantemente en el lado izquierdo, y el segundo estructura fetal rudimentaria y deforme, área cardíaca mal estructurada y latidos cardíacos solo arrítmicos y bradicárdicos, además de que no se definieron los órganos internos y todo se encontraba rodeado de linfangioma quístico grave con grandes quistes paravertebrales y un solo cordón umbilical con tres vasos. Lo descrito en la ecografía se corroboró con los resultados de la necropsia, lo cual se correspondió con la variedad Acardius mylacephalus.
Palabras clave: embarazo gemelar monoamniótico, embarazo gemelar monocigótico, transfusión feto-fetal, Acardius mylacephalus.
ABSTRACT
The case report of a 21 years primigravida woman is presented to whom a monoamniotic and monochorial twin pregnancy was detected in the echographic exam at the Genetic Provincial Center of Santiago de Cuba. The first fetus presented bilateral hydronephrosis predominantly in the left side, and the second rudimentary and deformed fetal structure, not well structured heart area and just arrhythmic and bradycardiac heart beats besides that the internal organs were not defined and everything was surrounded by severe cystic lymphangioma with big paravertebral cysts and a single umbilical cord with three vessels. Everything described in the echogram was corroborated with the results of the autopsy, which corresponded with Acardius mylacephalus variety.
Key words: monoamniotic twin pregnancy, monochorial twin pregnancy, fetus-fetal transfusion, Acardius mylacephalus.
INTRODUCCIÓN
La acardia fetal, también conocida como síndrome de transfusión feto-fetal o intergemelar, o síndrome de perfusión arterial reversa (TRAP, por sus siglas en inglés) constituye una de las malformaciones menos frecuentes descritas en la bibliografía médica, causada por un desequilibrio hemodinámico agudo o crónico, que se presenta de forma exclusiva en los embarazos gemelares monocigótico, que comparten anastomosis vasculares placentarias y se transfunde sangre de un gemelo a otro.1-4
Se presenta, así, un feto donador que es un gemelo estructuralmente sano, el que funciona como "bomba", y un feto receptor, que es un gemelo estructuralmente anormal, con un cuerpo rudimentario, denominado acárdico, que recibe sangre del gemelo "bomba". La evolución natural de esta entidad clínica implica una alta mortalidad intrauterina y neonatal, de hasta 55 % de los casos. La frecuencia de aparición varía de 1 cada 35 000 a 1 por cada 48 000 nacimientos y en 1 de cada 100 embarazos gemelares monocoriales.3-5
El primer caso se describió en 1533 como complicación de una gestación gemelar, pero la anomalía fue reconocida desde 1562 hasta 1720, cuando fue denominada como "monstruos sin cabeza". Los fetos acárdicos se han diagnosticado con la ecografía desde 1978, con la cual se observa una masa amorfa que recibe el flujo sanguíneo en forma reversa a través de la arteria umbilical, y además muestra que la secuencia TRAP no afecta todos los segmentos corporales al mismo tiempo, sino que algunos campos embrionarios pueden continuar su desarrollo.3
La conducta ante esta complicación continúa en la disyuntiva de si se debe intervenir o permanecer a la expectativa. La presentación de este caso clínico puede ser de interés para toda la comunidad científica, en especial en Cuba, donde el examen prenatal permite identificar tales malformaciones desde una temprana edad gestacional.
CASO CLÍNICO
Se describe el caso clínico de una gestante de 21 años de edad, primigesta, ama de casa, con color de la piel mestizo, procedencia urbana y antecedentes de salud y familiares de diabetes mellitus; quien fue remitida al Centro Provincial de Genética, localizado en el Hospital Infantil Docente Sur "Dr. Antonio María Béguez César" de Santiago de Cuba, a las 23 semanas de gestación, porque el diagnóstico ecográfico revelaba un tumor complejo, intraamniótico, en un embarazo supuestamente simple.
En el Centro se le realizó una nueva ecografía, la cual mostró un embarazo gemelar monoamniótico y monocigótico.
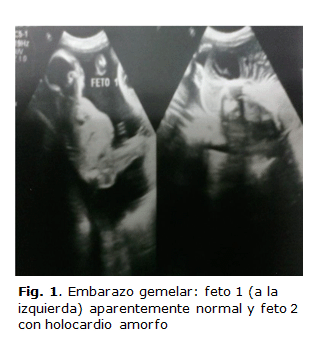
• Feto1: Biometría fetal acorde a la edad gestacional, que mostraba aumento de la ecogenicidad intestinal e hidronefrosis bilateral ligera. El resto de los parámetros se encontraban dentro de límites normales (figura 1).
• Feto 2: Estructura fetal rudimentaria y deforme, donde se definieron polo cefálico, esbozo de miembro inferior único y raquis rudimentarios, con una longitud cráneo-caudal de 84 mm para 14,6 semanas. No se definió área cardíaca bien estructurada, solo latidos cardíacos arrítmicos y bradicárdicos. Tampoco se observó el resto de los órganos internos, sino que todo estaba rodeado de linfangioma quístico grave con grandes quistes paravertebrales y un solo cordón umbilical con tres vasos (figura 2).
Teniendo en cuenta lo hallado, se diagnosticó un feto acárdico amorfo y se le informó a la madre y a los familiares en consulta especializada. Además se realizó asesoramiento genético específico y se explicó la posible causa, la evolución, el pronóstico y el riesgo elevado de complicaciones, tanto de prematuridad como de muerte fetal intrauterina del gemelo sano. Se citó a la madre para reevaluar el diagnóstico a los 15 días.
Se repitió la ecografía, en la cual se obtuvieron los mismos hallazgos, con las características descritas anteriormente; por lo que se le sugirió a la paciente la posibilidad de interrumpir voluntariamente el embarazo por causa genética. Ella aceptó y se le remitió al hospital ginecoobstétrico correspondiente para el procedimiento.
Resultados anatomopatológicos
El resultado de necropsia informó un embarazo gemelar monoamniótico y monocigótico.
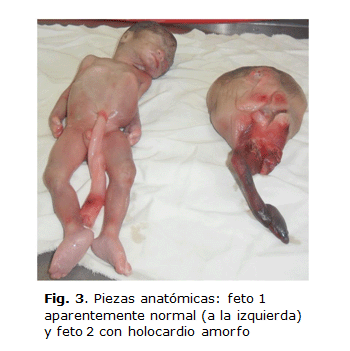
• Feto 1: Con hidronefrosis bilateral, predominantemente en el lado izquierdo. Peso de 400 gramos (figura 3).
• Feto 2: Masa tumoral amorfa con hipertricosis, que presentaba extremidad inferior rudimentaria a continuación de la región caudal izquierda, con 4 dedos. En la región cefálica mostraba aspecto multilobulado, que al corte semejaba al parénquima cerebral, licuado y con abundante contenido gelatinoso al romper los quistes. No se definieron estructuras del macizo facial, solo un orificio bucal rudimentario; tampoco presentaba ojos, nariz, orejas ni miembros superiores. Se esbozaba tejido cardíaco, escasos intestinos delgado y grueso, ambos riñones rudimentarios y columna vertebral normal desde la región dorsal (figura 4). No existía ni estómago ni pulmones. El cordón umbilical era normal y no se definía el sexo fetal. Peso de 200 g.
COMENTARIOS
Este caso se correspondió con el diagnóstico de holocardio amorfo de la variedad Acardius mylacephalus, que es una rareza de la gestación gemelar, vista únicamente en los embarazos gemelares monocigóticos. El último caso notificado en la provincia de Santiago de Cuba fue en el año 2007 y se trataba de la misma variedad, 6 con coincidencia de todo lo descrito en la ecografía con los resultados de la necropsia: presencia de polo cefálico rudimentario, con esqueleto axial y solo un miembro inferior mal desarrollado; ausencia de corazón de forma parcial y órganos internos mal desarrollados o casi omitidos. El que la paciente fuera primigesta y sin historia de embarazos múltiples, también concordó con un informe sobre esta anomalía.1
La fisiopatología de la enfermedad ha sido explicada por 2 vías: una es la vasculatura placentaria anormal que lleva a una circulación reversa, con alteración subsecuente del desarrollo cardíaco, y la otra se basa en una embriogénesis cardíaca anormal como episodio primario que lleva a un flujo retrógrado, lo cual puede deberse a una anormalidad cromosómica grave o a factores ambientales.3,4,6
Estas anomalías fetales se han clasificado morfológicamente, desde 1902, de la siguiente manera:5,7
- Acárdico acéfalo (65 %): Se caracteriza por ausencia de cabeza y órganos torácicos.
- Acárdico anceps (22 %): Se caracteriza por cabeza, cara y extremidades superiores parcialmente formadas.
- Acárdico acormus (5 %): Desarrollo solo de la cabeza, poco frecuente.
- Acárdico amorfo (8 %): Es una masa que conserva su estructura axial. Algunos agregan el nombre de Acardius mylacephalus al gemelo acárdico amorfo, que exhibe extremidad con algún grado de desarrollo.
Actualmente se utiliza un sistema de clasificación de Quintero 2 que establece 5 estadios progresivos: el estadio I, con diferencia de líquido amniótico entre los fetos; el II, se la suma la discordancia de los tamaños vesicales; en el III, en el estudio con ecografía Doppler está ausente el flujo diastólico o se encuentra reverso en la arteria umbilical del feto donante y/o flujo ausente o reverso en la contracción atrial del conducto venoso del feto receptor; el estadio IV presenta hidropesía fetal en cualquiera de los dos fetos; por último, el V existe muerte de un feto o de ambos. En el actual caso clínico la alteración congénita se encontraba en el estadio IV.
El corazón puede estar completamente ausente (holoacardius), ser una estructura rudimentaria (pseudoacardicus) o estar relativamente bien formado; en menos de 20 % de los gemelos acárdicos se reconoce tejido cardíaco.3 Así se presentó en este caso.
Los fetos acárdicos muestran una apariencia característica debido a que reciben sangre pobremente oxigenada a través de las arterias umbilicales. Las estructuras irrigadas por las arterias ilíacas y la aorta abdominal son relativamente bien formadas, mientras que el cuerpo superior y la cabeza reciben poca sangre oxigenada; usualmente los fetos no poseen cabeza, aunque se pueden encontrar esbozos rudimentarios, lo que lleva al sinónimo de esa entidad: acardio-acéfalo. De igual modo tienden a presentar gran edema de partes blandas e higroma quístico multiseptado en el dorso, además de extremidades superiores rudimentarias o la ausencia de esta; sin embargo, las extremidades inferiores pueden estar mejor formadas y el fémur habitualmente parece tener una configuración normal, como ocurrió en este estudio.
Resulta importante señalar que este trastorno genético se debe diagnosticar desde una temprana edad gestacional por medio de una ecografía, donde la de tipo Doppler tiene una función central y ofrece posibles terapéuticas. Por las características ecográficas, se debe realizar un diagnóstico diferencial con los teratomas placentarios o de cordón umbilical, de fetus in fetus y de muerte intrauterina de gemelo monocigótico gravemente malformado.
Esta malformación muestra una elevada mortalidad fetal y neonatal. Los gemelos acárdicos no son viables y la mortalidad perinatal del gemelo sano sin tratamiento excede 50 %, pues esta perfusión arterial retrógrada suele provocar alteraciones, como sobrecarga circulatoria, que puede aparecer junto con insuficiencia cardíaca, oligoamnios y anemia. El riesgo de prematuridad está presente como en cualquier embarazo gemelar.3,7,8
Hoy día en el mundo se tratan estos problemas según diferentes teorías, que incluyen terapia médica con digoxina a la madre para el control del fallo cardíaco del feto donador, amniocentesis descompresiva en los polihidramnios, aplicación de indometacina u otros antiinflamatorios no esteroideos a la madre para contribuir a la tocólisis y la disminución del polihidramnios. La terapia invasiva fetal, cuando falla el tratamiento médico, consiste en la reducción selectiva del feto acárdico bajo histerotomía y la continuación del embarazo del gemelo donador, pinzamiento de la circulación del feto acárdico bajo endoscopia o por histerotomía, embolización umbilical del feto acárdico con alcohol absoluto a través de cordocentesis guiada por ecografía, cauterización bipolar o uso de radiofrecuencia y electrocoagulación con láser del cordón umbilical del acárdico.4,6,8,9
REFERENCIAS BIBLIOGRÁFICAS
1. Cruz-Hernández María Mónica, Jaramillo-Valencia Juan Luis, Mejía-García Natasha, Gutiérrez-Marín Jorge Hernán, Sanín-Blair José Enrique. Acardius acormus: una presentación atípica en el embarazo gemelar. Revisión de la literatura. Rev Colomb Obstet Ginecol. 2009 [citado 20 Oct 2016]; 60(4): 382-6. Disponible en: http://www.scielo.org.co/scielo.php?script=sci_abstract&pid=S0034-74342009000400010&lng=es&nrm=iso
2. Molina M. Juan Carlos, García Flores Antonio, Torrico Aponte William Alexander, Vásquez Díaz Pedro Julián, Pardo Ramirez Pamela Ivette. Síndrome de transfusión feto-fetal: feto acárdico y acéfalo. Rev Méd. (Cochabamba) 2009 [citado 20 Oct 2016]; 20(30): 44-51. Disponible en: http://www.revistasbolivianas.org.bo/scielo.php?script=sci_arttext&pid=S2074-46092009000100009&lng=es&nrm=iso
3. Olaya-Contreras Mercedes, Derly Liseth Castro-Rojas. Feto acárdico: la malformación más grave en humanos. Patología. 2013 [citado 20 Oct 2016]; 51: 41-46. Disponible en: http://www.medigraphic.com/pdfs/patrevlat/rlp-2013/rlp131h.pdf
4. Ventura Laveriano W, Huertas Tacchino E, Limay Ríos A, Castillo Urquiaga W, Zárate Girao M, Díaz J, et al. Fetoscopia y coagulación bipolar selectiva en una gestación gemelar complicada con secuencia arterial reversa. A propósito del primer caso en el Perú. Rev Peru Ginecol Obstet. 2015 [citado 20 Oct 2016]; 61(1): 41-4. Disponible en: http://www.redalyc.org/pdf/3234/323438596007.pdf
5. Rivera Valdespino AC, García Jardon ME. Gestación gemelar con feto acárdico: Presentación de un caso. Rev Haban Cienc Méd. 2014 [citado 2 Mar 2016]; 13(4): 561-9. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1729 -519X2014000400007
6. Huerta-Saenz IH. Secuencia de perfusión arterial reversa en embarazo gemelar (TRAP) monocoriónico con feto acárdico. Rev Peru Ginecol Obstet. 2013 [citado 20 Oct 2016]; 59(3): 203-6. Disponible en: http://www.redalyc.org/pdf/3234/323429484011.pdf
7. Liens Garlobo I, Peña Reyes JM, Gómez Pérez H, Pons Porrata LM. Holocardio amorfo mielocéfalo. MEDISAN. 2007 [citado 20 Oct 2016]; 11(3). Disponible en: http://bvs.sld.cu/revistas/san/vol11_3_07/san13307.pdf
8. Gómez LF, Molina FS, Fresneda MD, Padilla MC. Secuencia TRAP: diagnóstico, opciones de tratamiento y experiencia propia. Diagn Prenat. 2012; 23: 160-6.
9. Otaño L, Meller CH, Aiello H. Avances en terapia fetal. Arch Argent Pediatr. 2013 [citado 17 Mar 2016]; 111(4): 332-44. Disponible en: http://www.scielo.org.ar/scielo.php?script=sci_arttext&pid=S0325-00752013000400013
Recibido: 13 de mayo de 2016.
Aprobado: 19 de octubre de 2017.
Valia Hernández Viel. Hospital Infantil Docente Sur "Dr. Antonio María Béguez César", avenida "24 de Febrero", nr 402, Santiago de Cuba, Cuba. Correo electrónico:valia.hernandez@infomed.sld.cu


{kind=link}
{kind=link}
{kind=link}
{kind=link}