










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La medicina moderna cuenta con poderosos recursos en el orden terapéutico y diagnóstico, aunque el método clínico sigue siendo el de mayor relevancia. En el siglo pasado y en el actual, el ejercicio de la profesión médica ha llegado a un plano superior, pues tanto la medicina celular como la molecular ya no constituyen un elemento abstracto sino un hecho, debido a su existencia y rápido desarrollo. Desgraciadamente, estos avances no están al alcance de todos los sistemas sanitarios del mundo, puesto que su aplicación es muy costosa.1,2
Ahora bien, entre las alteraciones innatas del metabolismo se encuentra la tirosinemia, de la cual existen varios subtipos que van desde la 1 hasta la 3. La tirosinemia de tipo 1 es un trastorno del metabolismo de los aminoácidos caracterizado por manifestaciones hepatorrenales y del sistema nervioso periférico, producto de una deficiencia de fumarilacetoacetato hidrolasa y fumarilacetoacetato isomerasa (últimas enzimas en la vía catabólica de la tirosina). Se estima, que existe una prevalencia de un caso por cada 2 millones de habitantes. Suele presentarse de forma aguda (neonatal o infantil) o crónica, después de los 2 años de vida.3,4
Son pocos los pacientes que presentan tirosinemia, escasos los profesionales que hablan sobre dicha afección y mucho menos quienes la estudian; tampoco abundan los laboratorios interesados en fabricar la medicación para tratar a estos pacientes.5,6 Actualmente existen métodos certeros de diagnóstico si se investigan los ácidos orgánicos urinarios y la presencia de succinilacetona. Zeybek et al7 coinciden con Guelbert, quien alerta a los pediatras para que, ante la simple duda o sospecha, realicen esas pruebas en lugar de suponer afecciones menores.
El tratamiento se basa en la administración de nitisinona, que interfiere el catabolismo y evita la producción de catabolitos tóxicos. A pesar del tratamiento, algunos pacientes presentan hepatoma con alfafetoproteína elevada y requieren trasplante de hígado. Aún, el diagnóstico precoz y el tratamiento dietético siguen siendo la única forma de evitar la muerte y mejorar, en lo posible, la calidad de vida de los pacientes con este trastorno innato.8
Caso clínico
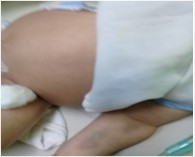
Se presenta el caso clínico de un paciente de 2 meses de edad, quien nació producto de un parto eutócico, a término y normopeso. Se alimentaba con lactancia materna exclusiva y tuvo antecedentes de 2 ingresos previos, uno, por anemia severa y otro, por catarro común. Fue hospitalizado en el Servicio de Terapia Intensiva del Hospital Infantil Sur “Dr. Antonio María Béguez César” por presentar edemas generalizados y ascitis (fig 1).
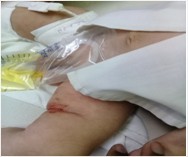
Evolutivamente presentó insuficiencia hepática aguda con heces acólicas, alteraciones sensoriales, íctero franco y hemorragias profusas (fig 2).
También mostró alteraciones humorales de la esfera hepática, tales como elevación de transaminasa glutámico pirúvica y glutámico oxalacética con valores de 115 y 90 U/L respectivamente; aumento de la fosfatasa alcalina hasta cifras de 410 U/L; bilirrubinas indirecta y directa con valores de 2 mg/dL y 16 mg/L, ambas aumentadas a expensas, sobre todo, de la fracción directa. También presentó hipoglucemia y coagulograma con deficiencia de factores dependientes de la vitamina K (II y VII). Continuó con deterioro progresivo del sensorio a pesar de que se indicó tratamiento específico para la insuficiencia hepática y se tomaron medidas drásticas para disminuir el ingreso de proteínas en la dieta.
Se realizaron estudios imagenológicos tales como ecografía abdominal, que mostró inicialmente hepatomegalia de 3 cm y de manera evolutiva, atrofia hepática, así como ausencia de alteraciones de las vías biliares. La tomografía abdominal corroboró los hallazgos de la ecografía. El estudio de la esfera renal arrojó valores de creatinina discretamente elevados de 134 mmol/L; el amoniaco fue de 180 mg/dL y la urea de 14,2 mmol/L. También se efectuaron pruebas metabólicas de orina con presencia de succinilacetona y se indicó confirmación en sangre, la cual también fue positiva, estos últimos exámenes confirmaron el diagnóstico de tirosinemia de tipo 1. A pesar de que el paciente fue tratado adecuadamente su evolución fue desfavorable.
Comentarios
Los pacientes con tirosinemia de tipo 1 nacen sanos, puesto que hasta el momento del parto su madre es la encargada de metabolizar las proteínas y lo hace bien, aunque sea portadora de una alteración metabólica. Cuando el niño comienza a alimentarse, las proteínas de la leche se degradan y liberan todos los aminoácidos, momento en el cual generalmente comienzan los síntomas de la enfermedad.9
Su diagnóstico se apoya en varios pilares básicos, que incluyen manifestaciones clínicas, antecedentes de muertes en la familia por cuadros de insuficiencia hepática inexplicables y apoyo de los exámenes complementarios sobre todo en el orden bioquímico y enzimático. La afectación hepática se pone de manifiesto por alteraciones en la coagulación, hiperbilirrubinemia e hiperamonemia moderadas, hipoglucemias y aminoaciduria. Los estudios enzimáticos son extremadamente caros, entre los cuales se encuentran: cultivo de linfocitos y fibroblastos que arroja la deficiencia de fumarilacetoacetato, pero el más útil y económico es la búsqueda cualitativa de hiperaminoaciduria en orina y sangre. La presencia de succinilacetona en muestras de orina y sangre es patognomónica de la tirosinemia de tipo 1.7-9,10
Este paciente nació a término y aparentemente sano. Como se recoge en la mayoría de los casos de tirosinemia hubo un periodo previo asintomático y de duración variable; luego comenzaron los síntomas propios de la enfermedad dados por elementos de insuficiencia hepática, hemorragias y unas pruebas metabólicas características que permitieron el diagnóstico de esta afección.
Si bien la tirosinemia de tipo 1 es una entidad observada con baja frecuencia, en la actualidad no existen elementos en el orden preventivo que permitan su detección más precoz, dado que no forma parte de las pruebas del tamiz neonatal en ningún país, lo que se justifica por su baja frecuencia. Solo la sospecha diagnóstica cuando un paciente presenta insuficiencia hepática en los primeros días o meses de vida puede indicar la presencia de esta afección, aunque dichos pacientes muestran características clínicas y humorales que pueden confundirse con otras enfermedades más frecuentes en esta etapa de la vida.

