










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome Klippel-Trenaunay es un padecimiento congénito raro, que fue descrito en 1900 por Klippel y Trenaunay. Este se manifiesta en la infancia o la adolescencia y su cuadro clínico consiste en angiomas cutáneos (hiperplasia, desarrollo excesivo de los tejidos y/o del tejido vascular sanguíneo) de color vino oporto, que pueden afectar a casi todas las partes del cuerpo, con inicio temprano de varicocidades e hipertrofia (aumento agudo de un órgano) de los tejidos blandos y óseos de un miembro. En 1907, Weber describió un paciente con los tres síntomas precedentes más una malformación arteriovenosa en la extremidad afectada.1
Este síndrome presenta una baja incidencia de 1 por cada 20 000 recién nacidos. Aunque hasta la fecha se desconoce su verdadera etiopatogenia, se considera la existencia de un defecto morfogenético que modifica la génesis angiovascular por un mecanismo de interferencia de la apoptosis durante la etapa temprana de la vida intrauterina; se refiere la intervención alterada de un factor antagonista vascular, la angiopoyetina II. Por otra parte, la hipertrofia de la extremidad afectada, con aumento del volumen y diámetro, así como de la longitud ósea, se le atribuye a un fenómeno de hipertensión venosa por trombosis, atresia del sistema venoso profundo o alteraciones en la regulación de la integración de canales vasculares.1,2
A pesar de que se desconoce su causa, se plantea la posibilidad de un defecto en la mutación del gen VG5Q que controla el crecimiento de los vasos sanguíneos. También se ha sugerido una posible herencia autosómica dominante, quizás una mutación somática de un gen letal que sobrevive por mosaicismo, lo cual puede ser más certero. Recientemente se ha comunicado un aumento en la transcripción del factor angiogénico VG5Q y la mutación del E133K, que fortalece la acción de esta molécula; sin embargo, se requieren más investigaciones para determinar la dinámica en la etiopatogenia de este trastorno.1,2
La principal complicación asociada al síndrome de Klippel-Trenaunay es la tromboflebitis que ocurre en 20-45 % de los pacientes y ocasiona embolismos pulmonares en 4-25 % de los casos. Las malformaciones pueden afectar, además, otros órganos, como el hígado, los intestinos, el bazo, los riñones. Cuando la malformación se ubica en los vasos de la columna vertebral, puede ocasionar escoliosis por lesión ósea.
Debe señalarse que el diagnóstico es esencialmente clínico y que el apoyo en las técnicas imagenológicas se basa principalmente, pero no de manera exclusiva, en la ecografía Doppler, la radiografía simple y la resonancia magnética nuclear. La arteriografía y angiografía son importantes para realizar los diagnósticos diferenciales.3,4
Caso clínico
Se describe el caso clínico de un neonato del sexo masculino, normopeso, producto de un embarazo a término, con índice de Apgar 9/9, asistido en el Hospital General Docente “Dr. Juan Bruno Zayas Alfonso” de Santiago De Cuba, sin antecedentes patológicos maternos ni familiares.
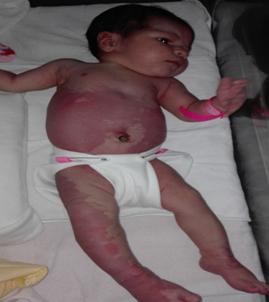
En el seguimiento genético de la etapa prenatal no se obtuvo diagnóstico alguno. Durante la realización de los cuidados inmediatos se observó macrocefalia, talla de 42 cm, asimetría de los miembros inferiores, marcada hipertrofia en el miembro inferior izquierdo, con macrodactilia; angiomas cutáneos en la porción inferior del tronco, el abdomen, la región glútea y las piernas (fig. 1). Se decidió ingresar al recién nacido en la Unidad de Cuidados Intensivos Neonatales para que un equipo multidisplinario evaluara su estado.
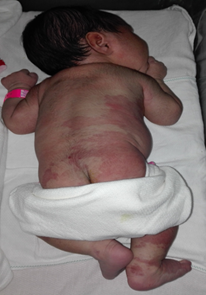
Fig. 1 Angiomas cutáneos en la porción inferior del tronco, el abdomen, la región glútea y las piernas.
Exámenes complementarios
Los resultados de los exámenes generales de laboratorio no revelaron anormalidad alguna.
La radiografía simple mostró la diferencia en el tamaño de ambos miembros pélvicos (fig. 2), atribuible a las partes blandas del lado derecho, sin daño óseo subyacente (fig. 3). Asimismo, el médico radiólogo determinó un incremento de la longitud de 0,8 cm de la extremidad prominente y una diferencia de altura de las crestas ilíacas de 3,1 cm.
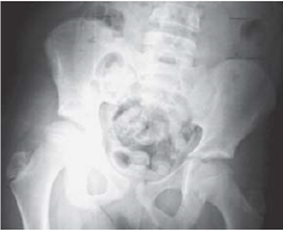
Diagnóstico y terapéutica aplicada
Se efectuó la interconsulta con los servicios de Ortopedia, Angiología Pediátrica y Genética, y en conjunto se diagnosticó un probable síndrome de Klippel-Trenaunay, de inicio temprano.
Por ello, se recomendó el uso de ropas adecuadas, según las anomalías musculo-esqueléticas, y vendaje de compresión media en el miembro afectado, que debía ser aplicado por las noches. De igual modo, se indicó mantener la observación en visitas médicas posteriores, evitando procedimientos intervencionistas de diagnóstico y tratamiento por la edad y la condición del paciente.
La familia fue orientada sobre las posibles complicaciones y los procedimientos posteriores, dado que en ocasiones es necesario el tratamiento quirúrgico si se presentan trastornos de coagulación de un miembro, donde se puede requerir hasta la amputación. También se emplean métodos quirúrgicos en las malformaciones venosas y la escleroterapia guiada por ecografía en las varicosidades y úlceras. Las pruebas más invasivas, pero muy importantes para el diagnóstico definitivo, como la resonancia magnética nuclear, la arteriografía y angiografía, se realizarán en edades más avanzadas.
En estos momentos ya el paciente es un lactante que no ha presentado complicaciones y en el que se mantiene un estricto control por parte de los especialistas en Angiología, Ortopedia y Genética Clínica.
Comentarios
El síndrome de Klippel-Trenaunay es una condición congénita rara caracterizada por angiomas de piel, con malformaciones arteriovenosas, además de hipertrofia de las partes blandas y óseas de la zona afectada. Es poco frecuente, con 1 por cada 20 000 nacidos vivos, lo que puede conducir a una evaluación o un tratamiento inadecuados, así como a un retraso en el diagnóstico, incluida la demora o no detección de posibles manifestaciones asociadas, que puede provocar limitación funcional en los enfermos, lo cual es potencialmente evitable. Las manifestaciones son proteiformes, por lo que la entidad clínica puede ser confundida con otros síndromes causantes de hipertrofia de miembros (síndrome de Proteus o de Parkes-Weber).5,6
Es una anormalidad vascular congénita de causa incierta, aunque se plantea la posibilidad de la mutación del gen VG5Q y del gen E133K, además de la relación con el gen para la proteína activadora de la guanosina trifosfatasa, cuya función es el control del crecimiento y la diferenciación celular; caracterizada por hemangioma plano, hipertrofia de huesos y tejidos blandos, que generalmente coinciden con el área de hemangioma y alteraciones del sistema venoso.6
La congestión venosa crónica puede llevar a la hiperpigmentación de la piel, tal como se evidencia en las imágenes del tronco y los miembros inferiores del paciente de este caso clínico. También pueden aparecer eczema, lipodermatoesclerosis, varicosidades, atrofia, flebectasia y, posteriormente, úlceras de la piel, las cuales son difíciles de curar. Es frecuente la alimentación de las displasias venosas por micro- o macrofístulas arteriovenosas, puesto que las macrofístulas extirpadas quirúrgicamente pueden presentar recidivas si son alimentadas por otras microfístulas.7
No siempre coinciden todas las características clínicas en un paciente; puede existir daño visceral con afección abdominal por el hemangioma ipsilateral, lo que incluye el colon, el hígado, el bazo, el yeyuno, los riñones; pero puede estar constituido solo por alteraciones cutáneas y melanosis de escalera bilateral sin daño visceral.6
Las complicaciones normalmente observadas en los pacientes con este trastorno son varias, entre ellas la celulitis por sobreinfección bacteriana, el dolor neuropático, la tromboflebitis y la úlcera varicosa. La celulitis en general no es grave y mejora con los antibióticos. Los dolores pueden llegar a ser intensos, por lo que se deben usar analgésicos durante tiempos prolongados. Igualmente, en todo paciente con dolor intenso de inicio súbito debe sospecharse la aparición de tromboflebitis, que, en general, son superficiales y limitadas, y se recomienda no ligar la vena. La trombosis venosa profunda puede presentarse hasta en 4 % de los casos.8,9
Aunque no existe cura para las personas que padecen el síndrome de Klippel-Trénaunay, los expertos de Mayo Clinic4 sugieren diferentes tratamientos para controlar los síntomas y evitar complicaciones, a saber:10
Terapia de compresión. Se envuelven las extremidades afectadas con vendas o prendas elásticas para prevenir la hinchazón, las venas varicosas y las úlceras de piel. Se pueden usar dispositivos de compresión neumática intermitente, que son fundas para piernas o brazos que se inflan y desinflan automáticamente a intervalos fijos.
Cuidado del estilo de vida. Los zapatos ortopédicos pueden mejorar la salud y la función física.
Fisioterapia. Los masajes, la compresión y estimulación del uso adecuado de las extremidades puede ayudar a aliviar la hinchazón de los brazos o las piernas (linfedema) y la hinchazón de los vasos sanguíneos.
Epifisiodesis. Es un procedimiento ortopédico que puede detener el crecimiento excesivo de las extremidades inferiores.
Embolización. Este procedimiento, que se realiza mediante pequeños catéteres en las venas o las arterias, bloquea el flujo sanguíneo a determinados vasos sanguíneos.
Terapia con láser. Este procedimiento puede aclarar o eliminar los hemangiomas planos de la piel.
Ablación por láser o por radiofrecuencia de las venas. Este procedimiento mínimamente invasivo se utiliza para bloquear venas anormales.
Escleroterapia. El médico te inyecta una solución en la vena, lo que crea tejido cicatricial que ayuda a bloquear la vena.
Colocación de un filtro en la vena cava. Este procedimiento evita que los coágulos sanguíneos se trasladen a los pulmones.
Tratamiento quirúrgico. En algunos casos es posible el beneficio con la extracción o la reconstrucción de las venas afectadas, así como con la corrección del crecimiento excesivo de los huesos.
Además, es posible que se necesite tratamiento específico si aparecieran complicaciones como sangrado, infección, coágulos sanguíneos o úlceras cutáneas.
Hasta el momento el paciente, que ya es un lactante de más de 3 meses, no ha presentado complicaciones y solo ha necesitado medidas terapéuticas generales.

