










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicentro Electrónica
versión On-line ISSN 1029-3043
Medicentro Electrónica vol.19 no.4 Santa Clara set.-dic. 2015
INFORME DE CASO
Síndrome de Cornelia de Lange. Presentación de un paciente
Cornelia de Lange syndrome. A patient report
MSc. Dra. Elayne Esther Santana Hernández1, MSc. Dr. Víctor Jesús Tamayo Chang2, MSc. Lic. Onelis Pupo Zalazar3
1. Especialista de Primer y Segundo Grados en Medicina General Integral y en Genética Clínica. Master en Atención Integral al Niño. Asistente. Investigador agregado. Centro Provincial de Genética Médica. Holguín. Cuba. Correo electrónico: esantana@hpuh.hlg.sld.cu
2. Especialista de Primer y Segundo Grados en Genética Clínica. Master en Atención Integral al Niño. Profesor Auxiliar. Investigador agregado. Centro Provincial de Genética Médica. Holguín. Cuba. Correo electrónico: vtamayo@hpuh.hlg.sld.cu
3. Licenciada en Enfermería. Master en asesoramiento Genético. Instructora. Centro Municipal de Genética. Moa, Holguín. Cuba. Correo electrónico: onelisp@moa.hlg.sld.cu
El primer caso de síndrome Brachmann de Lange o Cornelia de Lange (SCDL) fue descrito por Brachmann en 1916,1 y en 1933 Lange presentó pacientes con similares características fenotípicas.1,2 Esta enfermedad genética es poco frecuente; se presenta con una prevalencia y una incidencia que difiere según las poblaciones estudiadas. En España, la prevalencia es de 0,97 por 100 000, y en Estados Unidos, de 1 por 10 000.2
El diagnóstico es fundamentalmente clínico; se basa en el reconocimiento de los rasgos fenotípicos faciales característicos, unido a las malformaciones en las extremidades; esto puede acompañarse de fallo del crecimiento pre- y posnatal, así como discapacidad intelectual de grado variable.2
Los hallazgos distintivos faciales incluyen: cejas anchas bien pobladas, curvadas y unidas (sinofris), pestañas largas, orificios nasales en anteversión, micrognatia, microbraquicefalia en ocasiones, implantación baja del cabello y de las orejas, hipertricosis, puente nasal bajo, filtro largo, y boca en forma de carpa con labios finos y prognatismo maxilar.2-4
Las anomalías de los miembros superiores pueden ser mayores o menores, según el defecto que produzcan: micromelia, focomelia, implantación proximal de los pulgares, oligodactilia, agenesia o hipoplasia del cúbito, sindactilia y línea simiana. El llanto es típico y algunos pacientes pueden presentar convulsiones. Se han observado anomalías oculares, esqueléticas, cardíacas, digestivas, neurosensoriales, cutáneas, genitales y endocrinas.5 El síndrome de Cornelia se asocia, además, con alteraciones del comportamiento, tipo autista o hiperactividad, además de retraso mental de grado variable.6
La variabilidad clínica de este síndrome permite el reconocimiento de las diferentes formas por el grado de las malformaciones de los miembros: la forma grave clásica tipo I y la forma leve tipo II, que se presentan en el 20 % de los casos y pueden permanecer irreconocibles hasta la edad adulta. Casi todos los casos del síndrome de Cornelia son esporádicos, aunque se describen familias con patrón de herencia autosómica dominante.4-6
En la forma clásica tipo I del SCDL, los pacientes presentan restricción del crecimiento intrauterino (CIUR), retardo psicomotor moderado a profundo y malformaciones mayores que producen discapacidades graves o la muerte.7 En el tipo II o forma leve, las características faciales y anormalidades esqueléticas menores son similares a las del tipo I; sin embargo, los problemas funcionales del síndrome pueden desarrollarse en el tiempo o expresarse parcialmente. Por lo general, hay un retardo psicomotor leve a limítrofe, una restricción muy marcada del crecimiento pre- y posnatal y ausencia de malformaciones mayores.8,9
En este informe, se describe a un paciente con el síndrome de Cornelia de Lange que presenta malformaciones mayores en sus miembros, lo que posibilitó realizar el diagnóstico clínico de la enfermedad por las características fenotípicas, a los 20 días de nacido en la consulta de Genética Clínica.
Presentación del paciente
Se trata de un lactante que fue llevado a la consulta de Genética a los 21 días de nacido, con malformaciones graves en las extremidades y anomalías faciales características de esta enfermedad, con antecedentes prenatales de una madre primigesta de 21 años, con embarazo de bajo riesgo genético y moderado riesgo obstétrico por desnutrición materna.
En la etapa prenatal se le realizaron dos ultrasonidos, en los cuales no se diagnosticaron las alteraciones de los miembros, y solo se encontró líquido amniótico disminuido.
Antecedentes familiares: No se describen familiares con defectos de extremidades ni discapacidad mental.
Antecedentes perinatales: El parto se le presentó a las 39 semanas; el trabajo de parto duró seis horas y el producto fue un recién nacido masculino que pesó 2 600 g, talla: 48 cm; perímetro cefálico: 32 cm; perímetro torácico: 33 cm, y malformaciones en manos y pies.
Entre las características clínicas faciales, se encontraron: cabello de implantación baja anterior, hirsutismo marcado, cejas muy pobladas y unidas en la línea media (sinofris), pestañas largas, puente nasal ancho y bajo, filtro largo, boca «en tienda de campaña» con labios delgados. En el momento del nacimiento se observó ectrodactilia, sin poderse determinar los dedos faltantes por no estar todos los puntos de osificación para definir este dato (Figuras 1,2).
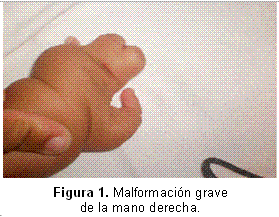
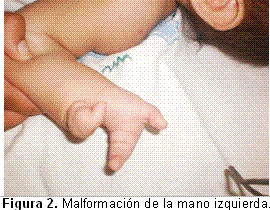
Respecto a los miembros inferiores, también están afectados ambos pies, pero en grado menor, ya que solo presentan sindactilia bilateral del segundo al quinto dedos, como se advierte en la Figura 3.

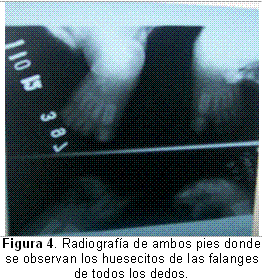
En la Figura 4 se pueden observar las falanges de todos los dedos, por lo que se confirma que tiene una sindactilia membranosa.
Después de examinar este paciente, de realizar los estudios requeridos y precisar bien el fenotipo por las características faciales de las cejas gruesas con sinofris y las malformaciones de los miembros, tanto en manos como en pies de manera marcada, se diagnostica como un afectado de SCDL tipo I o forma clásica.
Comentario
Este caso constituye el primero que se presenta con la forma más grave o tipo I en la consulta de Genética Clínica del Centro de Genética de la provincia de Holguín, Cuba. A pesar de las malformaciones de los miembros muy marcadas, el desarrollo pondoestatural y psicomotor se encuentra en correspondencia con su edad, aspecto que difiere de lo descrito por otros autores.1-3 Aunque en Cuba no existen datos sobre la incidencia y prevalencia de este síndrome, en la literatura se informa una prevalencia global de 0,6 por 100 000 nacidos vivos.2
Los estudios citogenéticos son normales en la mayoría de los pacientes, aunque se han descrito múltiples casos con alteraciones en el cromosoma 3, algunos con duplicación especifica del área q21-qter y otros con deleciones del brazo largo. También se conoce de familias que presentan una translocación equilibrada con puntos de quiebre en 3q26.3 y 17q23.1, por lo cual algunos autores proponen que el gen causante del SCDL se puede localizar en la región 3 q26.3. Hay informes de otras translocaciones equilibradas de novo, que se asocian con el SCDL, como 5p13.1 y 13q12.1 y que dañarían otros genes diferentes a los del cromosoma 3.4-6
El diagnóstico diferencial, en los casos con retardo en el crecimiento intrauterino y alteraciones en las extremidades superiores, incluye los síndromes de Grebbe, Roberts, Child y TAR, ninguno de los cuales presenta los hallazgos faciales característicos del SCDL.6-8 La sumatoria de los hallazgos en el examen físico, el cariotipo y el antecedente materno de no haber existido exposición alguna a tóxicos, permite clasificarlo como SCDL tipo I o forma clásica.9
El diagnóstico ultrasonográfico in utero de las malformaciones de las extremidades en este paciente hubiese ayudado a pensar en este síndrome. Este caso constituye un fallo del programa prenatal en la detección de malformaciones congénitas, lo que motivó realizar una estrategia de capacitación de radiólogos incorporados a los servicios de diagnóstico prenatal, para que malformaciones como estas sean detectadas oportunamente; asimismo, condujo a la búsqueda cuidadosa de los cuatro miembros en los diferentes ultrasonidos prenatales. Estos hallazgos no solo permiten el diagnóstico de SCDL, sino que admiten establecer el pronóstico del feto y planear posibles intervenciones in utero o neonatales.
El diagnóstico temprano de esta enfermedad posibilita iniciar una rápida atención integral al paciente, que incluye asesoramiento familiar, ayuda médica y estimulación psicomotora apropiada, para un mejor desempeño del niño.
REFERENCIAS BIBLIOGRÁFICAS
1. Kline AD, Calof AL, Schaaf CA, Krantz ID, Jyonouchi S, Yokomori K, et al. Cornelia de Lange syndrome: Further delineation of phenotype, cohesin biology and educational focus, 5th. Biennial Scientific and Educational Symposium abstracts. Am J Med Genet A [internet]. 2014 Jun. [citado 20 oct. 2014];164(6):[aprox. 10 p.]. Disponible en:http://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.36417 /abstract;jsessionid=BBD8B16CFBB29A999D20F6B1BF96A518.f02t01
2. Pavlidis E, Cantalupo G, Bianchi S, Piccolo B, Pisani F. Epileptic features in Cornelia de Lange syndrome: Case report and literature review. Brain Dev [internet]. 2014 Jan. 22 [citado 20 feb. 2014];36(10):[aprox. 7 p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/24461912
3. Dave U, Shetty D. Mutational Screening and Prenatal Diagnosis in Cornelia de Lange syndrome. J Obstet Gynaecol India [internet]. 2013 Sep. 29 [citado 20 feb. 2014];64(1):[aprox. 5 p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3931894/
4. Teresa-Rodrigo ME, Eckhold J, Puisac B, Dalski A, Gil-Rodríguez MC, Braunholz D, et al. Functional Characterization of NIPBL Physiological Splice Variants and Eight Splicing Mutations in Patients with Cornelia de Lange Syndrome. Int J Mol Sci [internet]. 2014 Jun. 10 [citado 30 nov. 2014];15(6):[aprox. 14 p.]. Disponible en:http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4100155/
5. Zuin J, Franke V, van Ijcken WF, van der Sloot A, Krantz ID, van der Reijden MI, et al. A cohesin-independent role for NIPBL at promoters provides insights in CdLS. PLoS Genet [internet]. 2014 Feb. 13 [citado 20 jun. 2014];10(2):[aprox. 13 p.]. Disponible en:http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3923681/
6. Kaiser FJ, Ansari M, Braunholz D, Gil-Rodríguez MC, Decroos C, Wilde JJ, et al. Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Hum Mol Genet [internet]. 2014 Jan. 8 [citado 20 jun. 2014];23(11):[aprox. 12 p.]. Disponible en: http://hmg.oxfordjournals.org/content/23/11/2888.long
7. Barbero JL. Genetic basis of cohesinopathies. Appl Clin Genet [internet]. 2013 May 1 [citado 20 feb. 2014];6:[aprox. 9 p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3711096/
8. Gervasini C, Parenti I, Picinelli C, Azzollini J, Masciadri M, Cereda A, et al. Molecular characterization of a mosaic NIPBL deletion in a Cornelia de Lange patient with severe phenotype. Eur J Med Genet [internet]. 2013 Mar. [citado 20 feb. 2014]; 56(3):[aprox. 6 p.]. Disponible en: http://linkinghub.elsevier.com/retrieve/pii /S1769-7212(13)00003-7
9. Marchisio P, Selicorni A, Bianchini S, Milani D, Baggi E, Cerutti M, et al. Audiological findings, genotype and clinical severity score in Cornelia de Lange syndrome. Int J Pediatr Otorhinolaryngol [internet]. 2014 Jul. [citado 30 oct. 2014];78(7):[aprox. 4 p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/24774220
Recibido: 23 de noviembre de 2014
Aprobado: 20 de febrero de 2015
MSc. Dra. Elayne Esther Santana Hernández. Especialista de Primer y Segundo Grados en Medicina General Integral y en Genética Clínica. Master en Atención Integral al Niño. Asistente. Investigador agregado. Centro Provincial de Genética Médica Holguín. Cuba. Correo electrónico: esantana@hpuh.hlg.sld.cu

