










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMedicentro Electrónica
versión On-line ISSN 1029-3043
Medicentro Electrónica vol.21 no.4 Santa Clara oct.-dic. 2017
INFORME DE CASO
Hallazgo cromosómico asociado a hipotiroidismo congénito: presentación de una paciente
Chromosomal finding associated with a congenital hypothyroidism: a case report
Elayne Ester Santana Hernández1, Víctor Jesús Tamayo Chang1, Elena Gertrudis Rodríguez Font2
1. Centro Provincial de Genética Médica de Holguín. Cuba. Correo electrónico: elsantana@infomed.sld.cu
2. Hospital Pediátrico Universitario de Holguín. Cuba. Correo electrónico: efont@infomed.sld.cu
RESUMEN
Es llevada al Centro Provincial de Genética Médica de Holguín, una paciente de ocho años de edad por dismorfias faciales, en las extremidades y discapacidad intelectual de moderada a grave. Se realizó el cariotipo en sangre periférica utilizando la microtécnica de cultivo de linfocitos. Las 22 metafases analizadas mostraron el hallazgo cromosómico de un cariotipo: 46, XX, del (18) (pter 11.1), en una paciente con deleción terminal en el brazo corto del cromosoma 18, asociado a hipotiroidismo congénito. El objetivo de este trabajo es describir un hallazgo citogenético asociado con una enfermedad genética metabólica que agrava su cuadro clínico. Se considera importante realizar el diagnóstico precoz de estas enfermedades genéticas para realizar el tratamiento oportuno, la estimulación temprana y brindar un adecuado asesoramiento genético a los familiares.
DeCS: deleción cromosómica, monosomía/genética.
ABSTRACT
An eight-year-old female patient was taken to the Provincial Center of Medical Genetics from Holguin due to her facial and limb dysmorphias, and moderate to severe intellectual disability. The karyotype was performed in peripheral blood using lymphocyte culture micro-technique. The 22 analyzed metaphases showed the chromosomal finding of a karyotype: 46, XX, der (18), (pter 11.1), in a patient with terminal deletion on the short arm of chromosome 18, associated with a congenital hypothyroidism. The aim of this study is to describe a cytogenetic finding associated with a genetic metabolic disease, aggravating its clinical manifestations. It is considered important to carry out the early diagnosis of these genetic diseases in order to provide timely treatment, early stimulation, as well as, an adequate genetic counseling to family members.
DeCS: chromosome deletion, monosomy/genetic.
Las aberraciones cromosómicas de estructura se caracterizan porque siempre existen puntos de ruptura del ADN que determinan rearreglos importantes. En las no balanceadas, el individuo afectado expresa en su fenotipo alguna anormalidad, cuya gravedad depende del cromosoma involucrado y la magnitud del defecto.1,2 Las deleciones son pérdidas de un fragmento del cromosoma; puede ser terminal, si el fragmento distal es el que se pierde, como lo ocurrido en este caso, en que se pierde el extremo terminal del brazo corto del cromosoma 18.3,4
La deleción del brazo corto del cromosoma 18 (18p) se informa por primera vez en 1963 por el genetista francés Jean de Grouchy y colaboradores, y constituye el primer ejemplo de monosomía parcial compatible con la vida. La literatura ha informado, aproximadamente, unos 180 casos, con una incidencia estimada aproximada 1: 50 000 recién nacidos vivos. Con gran variabilidad en sus fenotipos, los más frecuentes son la discapacidad intelectual de moderada a grave, la baja talla, la ptosis palpebral, el epicanto y las orejas prominentes.5-7
Se describe una proporción mayor de féminas afectadas en relación con los varones de 3/2. En la mayoría de los casos (85 %), se trata de deleciones nuevas, es decir, de novo, que aparecen en una fase precoz del desarrollo embrionario; es rara la transmisión familiar de la deleción (18p) y solo el 10 % son heredadas por translocaciones de los padres.8,9
El diagnóstico precoz de esta aberración cromosómica y el seguimiento multidisciplinario favorece el desarrollo de estos afectados, con una estimulación oportuna, para lograr una adecuada incorporación social. Es muy importante el papel del genetista clínico, que brinda la información a través del asesoramiento genético a las familias que lo requieran, para su próxima descendencia.10
Por ser esta aberración cromosómica poco frecuente y no sospecharse, mucho menos cuando se asocia con otra enfermedad que puede explicar la discapacidad intelectual en los casos de abandono del tratamiento, como resulta en el hipotiroidismo congénito no tratado adecuadamente, se consideró importante el estudio y presentación de esta paciente.
Presentación del paciente
Paciente femenina de ocho años, que nació por parto eutócico a las 39 semanas, con peso de 3080 gramos, talla 50 cm, Apgar 8-9; al realizar el examen físico, el neonatólogo describe a un recién nacido hipotónico, con varias dismorfias faciales y en la extremidades. No se refieren antecedentes prenatales ni perinatales de interés.
A la consulta de Genética Clínica asiste esta paciente acompañada de su madre, porque se le diagnóstico discapacidad cognitiva importante. Después de examinarla, se observan dismorfias faciales y en las extremidades, muy pálidas y edematosas, acompañadas de movimientos lentos y marcha con dificultad, unido a su discapacidad intelectual, que dificulta el examen.
Se le indica cariotipo en sangre periférica y se interconsulta de inmediato con las especialidades de Pediatría y Endocrinología; se le realiza diagnóstico clínico de hipotiroidismo descompensado, y se decide ingresarla para su mejor estudio y tratamiento.
En la sala, se realiza hemoglobina y se le diagnostica anemia (8,7g/l); se realiza TSH: 72 U/I, T4: 37 U/I, los cuales resultan alterados, y se revisan los registros de tamizaje neonatal. Según los antecedentes patológicos personales y los registros de tamizaje, se conoce que padece un hipotiroidismo congénito con tamizaje neonatal en 16 U/I, que se le suministró tratamiento al nacimiento y que, después de dos años, se ausentaron de la consulta y no mantuvo la terapia indicada.
El estudio de cariotipo en 22 metafases resultó: 46, XX, del (18)(pter 11.1), que consiste en una deleción completa de la porción terminal del brazo corto del cromosoma 18, una monosomía parcial 18p.
Se realiza el estudio cromosómico a ambos padres para investigar si ellos padecían una translocación balanceada o no. Los cariotipos materno y paterno fueron normales, por lo que se consideró una deleción nueva lo ocurrido a esta niña.
No se obtuvo ningún antecedente patológico familiar de interés. Se le pide consentimiento informado a los padres para tomarle fotos y ser publicadas en revistas médicas.
Entre las características faciales de la paciente, se encontraron: frente amplia, implantación alta del cabello, pelo grueso, cejas muy pobladas, hendiduras palpebrales hacia abajo, epicanto inverso e hipertelorismo, hipoplasia del tercio medio de la cara, filtrum largo, labio inferior grueso y superior muy fino, como se puede apreciar en la figura 1.

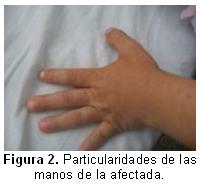
En las manos se observan uñas hipoplásicas con la falange distal muy corta, braquidactílea ligera, primer dedo muy grueso, piel redundante con hiperqueratosis, como se puede observar en la figura 2.
Comentario
Esta aberración cromosómica tiene expresividad variable, y muestra gran heterogeneidad clínica; se aprecian rasgos dismórficos, como cara redonda plana y sin expresión, el puente nasal es plano y ancho, las fisuras palpebrales hacia abajo con pliegues horizontales epicánticos; pueden encontrarse también estrabismo y ptosis de los párpados. La boca es ancha, el surco naso labial es bastante corto en ocasiones, aunque por lo general se pueden observar también filtrum largo que sobresale, con un labio superior fino, que llega a ser plano y el inferior grueso, a menudo invertido. Los dientes establecidos irregulares son de mala calidad, con caries significativas, los incisivos laterales aveces faltan, el paladar puede ser alto y el mentón, pequeño.1 Las orejas son grandes, con antehélix hipoplásico, y a menudo de ajuste bajo. Las manos son anchas y cortas con falanges de anchura decreciente.1-4 El cuello es corto y el tórax ancho con pezones muy separados (teletelia). Por lo general, presentan talla baja y siempre discapacidad intelectual variable; en esta enferma, la discapacidad intelectual es mayor, por presentar hipotiroidismo congénito, con abandono del tratamiento desde los dos años de edad.5,6
El análisis citogenético se realizó con el objetivo de llegar a un diagnóstico definitivo. El riesgo de recurrencia para los hermanos es bajo en deleciones que no son heredadas, pero es significativo si un reordenamiento de los padres está presente.7,8 La monosomía 18p puede detectarse de forma prenatal mediante amniocentesis o muestreo de vellosidades coriónicas, en estudio citogenético. El diagnóstico diferencial puede incluir una amplia serie de síndromes, con talla baja y deficiencia intelectual leve.9,10
No existe un tratamiento específico ni curativo para las aberraciones cromosómicas; sin embargo, resulta importante el diagnóstico temprano para su desarrollo posnatal, para comenzar una estimulación precoz; esto incluye terapia del lenguaje y los programas educativos tempranos, que pueden ayudar a mejorar el rendimiento. Resulta valiosa, para los familiares y la sociedad, la adecuada incorporación educativa y social de estos afectados. A excepción de los pacientes con malformaciones cerebrales graves, la esperanza de vida no parece reducirse significativamente.
Conflicto de intereses
Los autores declaran que no existen conflictos de intereses en el presente artículo.
REFERENCIAS BIBLIOGRÁFICAS
1. Shi S, Guo L, Zha Q, Shi Z, Yang Y. Genotype and phenotype analysis of a child with partial 18q deletion syndrome. Zhonghua Yi Xue Yi Chuan Xue Za Zhi [internet]. 2017 Aug. 10 [citado 12 sep. 2017];34(4):[aprox. 4 p.]. Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/28777861
2. Xu LJ, Wu LX, Yuan Q, Lv ZG, Jiang XY. A case of 18p deletion syndrome after blepharoplasty. Int Med Case Rep J [internet]. 2017 Jan. 12 [citado 12 sep. 2017];10:[aprox. 4 p.]. Disponible en: https://www.dovepress.com/a-case-of-18p-deletion-syndrome-after-blepharoplasty-peer-reviewed-fulltext -article-IMCRJ
3. Pachajoa H. Síndrome por deleción 18p diagnosticado por array de hibridación genómica comparada. Presentación de un caso con fenotipo leve. Arch Argent Pediatr [internet]. 2016 dic. 1 [citado 12 sep. 2017];114(6):[aprox. 2 p.]. Disponible en: http://www.sap.org.ar/docs/publicaciones/archivosarg/2016 /v114n6a30.pdf
4. Qiang YY, Zhang N, Gao YM. A case of whole arm deletion of chromosome 18p. Zhonghua Er Ke Za Zhi [internet]. 2016 Jul. [citado 12 sep. 2017];54(7):[aprox. 1 p.]. Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/27412746
5. Mahjoubi F, Razazian F, Torabi R. Clinical Features of a Case with 46,XX,del(18)(p11.1p11.3). Genet Couns [internet]. 2015 [citado 12 sep. 2017];26(3):[aprox. 4 p.]. Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/26625672
6. Hasi-Zogaj M, Sebold C, Heard P, Carter E, Soileau B, Hill A, et al. A review of 18p deletions. Am J Med Genet C Semin Med Genet [internet]. 2015 Sep. [citado 12 sep. 2017];169(3):[aprox. 14 p.]. Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31445/full
7. Wei J, Xie Y, He W, Liu W, Jian W, Chen M, et al. Clinical outcome: a monosomy 18p is better than a tetrasomy 18p. Cytogenet Genome Res [internet]. 2014 [citado 12 sep. 2017];144(4):[aprox. 5 p.]. Disponible en: https://www.karger.com/Article/FullText/371461
8. Sebold C, Soileau B, Heard P, Carter E, O'Donnell L, Hale DE, et al. Whole arm deletions of 18p: medical and developmental effects. Am J Med Genet A [internet]. 2015 Jan. 14 [citado 12 sep. 2017];167(2):[aprox. 11 p.]. Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.36880/full
9. Quiñones Maza O, Quintana Aguilar J, Méndez Rosado LA, Barrios Mesa A, Suárez Mayedo U, García M, del Sol M. Frecuencias de reordenamientos cromosómicos estructurales acorde a las indicaciones para estudios citogenéticos prenatales y postnatales. Rev Cubana Genét Comunit. 2010;4(3):36-42.
10. Willoughby BL, Favero M, Mochida GH, Braaten EB. Neuropsychological Function in a Child with 18p Deletion Syndrome: A Case Report. Cogn Behav Neurol [internet]. 2014 Sep. [citado 12 sep. 2017];(3):[aprox. 6 p.]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4176699/
Recibido: 14 de enero de 2017
Aprobado: 10 de mayo de 2017
Elayne Ester Santana Hernández. Centro Provincial de Genética Médica de Holguín. Cuba. Correo electrónico: elsantana@infomed.sld.cu

