










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkCorreo Científico Médico
versión On-line ISSN 1560-4381
ccm vol.20 no.1 Holguín ene.-mar. 2016
ARTÍCULO DE REVISIÓN
Mecanismos etiopatogénicos en la esclerosis sistémica
Ethiopathogenic Mechanisms in the Systemic Sclerosis
Susel E Remedios Batista 1, Ernesto Montada Cedeño 2, Eduardo Del Campo Avilés 3, Liliana Torres Pérez 4, Alexander Fernández Portelles 5, Orlando S. Paneque Landrove 6
1. Especialista en Primer Grado en Medicina General Integral y Reumatología. Asistente. Hospital Clínico Quirúrgico Carlos Font. Banes. Holguín. Cuba.
2. Especialista en Primer Grado en Medicina General Integral. Residente tercer año de Reumatología Hospital Clínico Quirúrgico Lucia Iñiguez Landín. Holguín. Cuba.
3. Especialista en Primer Grado en Reumatología. Instructor. Hospital Clínico Quirúrgico Lucia Iñiguez Landín. Holguín. Cuba.
4. Especialista en Primer Grado en Reumatología. Asistente. Hospital Clínico Quirúrgico Lucia Iñiguez Landín. Holguín. Cuba.
5. Especialista en Primer Grado en Medicina General Integral y Reumatología. Asistente. Hospital Clínico Quirúrgico Lucia Iñiguez Landín. Holguín. Cuba.
6. Especialista en Primer Grado en Reumatología. Asistente. Hospital Pediátrico Octavio de la Concepción y de la Pedraja. Holguín. Cuba.
RESUMEN
Se realizó un compendio de los principales factores etiopatogénicos causantes de la expresividad clínica de la esclerosis sistémica, aún no bien establecida, la asociación genética, herencia, factores ambientales y respuesta inflamatoria, su relación con infecciones endógenas comportándose de manera especial en otras enfermedades reumáticas inflamatorias. El objetivo del artículo fue describir los elementos de la etiopatogenia que distinguen a esta enfermedad del colágeno. Los mecanismos etiopatogénicos basados en anormalidades moleculares, pueden ser correctas en la esclerosis sistémica. Su espectro clínico heterogéneo responde a una etiopatogenia distinta en cada individuo, así como, a eventos complejos que pueden ocurrir en fases iniciales de la enfermedad y no a una patogenia única.
Palabras clave: esclerosis sistémica, microquimerismo, mimetismo molecular, productos eskleros, proteínas Smad
ABSTRACT
A summary of the main ethiopathogenic factors that cause the clinical expressiveness of the Systemic Sclerosis, still as soon as established, the genetic association, inheritance, environmental factors and inflammatory response, their relationship with endogenous infections behaving from a special way to other inflammatory rheumatic illnesses. The objective of this article was to describe the ethiopathogenic elements that distinguish this illness of the collagen. The ethiopathogenic mechanisms based on molecular abnormalities can be correct in the systemic sclerosis. Its heterogenic clinical spectrum responds to a different etiopathogeny in each individual, as well as to complex events that can happen in initial phases of the illness and not to a unique pathogenic.
Keywords: systemic sclerosis, microkimerism, molecular mimetism, eskleros products, Smad proteins.
INTRODUCCIÓN
La esclerosis sistémica (ES) 1 es una enfermedad extremadamente compleja, sus formas de presentación son diversas. Su inicio puede ser insidioso, en forma de dolores generalizados, rigidez, fatigabilidad y pérdida de peso. La historia clínica, sin embargo, más frecuente es la de un paciente que presenta fenómeno de Raynaud (palidez, rubicundez y cianosis en zonas distales) desde unos pocos meses a muchos años, al tiempo que nota un progresivo endurecimiento de la piel, que puede ser generalizado o limitado a las partes acras del cuerpo. El fenómeno de Raynaud es la primera manifestación en casi el 90% de los casos, hasta el punto de que su ausencia introduce dudas en el diagnóstico. A veces, en un número no despreciable de pacientes, el endurecimiento cutáneo está precedido por varios años de evolución del mismo 2.
Así, pues, ante todo fenómeno de Raynaud es obligado establecer si se trata de un evento primario o si puede ser la primera manifestación de la enfermedad. La capilaroscopia es la técnica de referencia para distinguir el fenómeno de Raynaud, primario del secundario 3. Con pocas excepciones, la presencia de alteraciones capilaroscópicas (sobre todo dilataciones y hemorragias) es indicativa de que el paciente desarrollará, con el tiempo, una conectivopatía, que en la mayoría de las ocasiones será una ES. También ayuda a sospechar y puede ser el primer signo de la enfermedad cuando se manifiesta cerca de los 40 años, si es grave y ocasiona úlceras necróticas digitales, si en la capilaroscopia del lecho ungueal, como ya se ha dicho, se observan alteraciones de la microcirculación y si los anticuerpos antinucleares son positivo 4, 5.
La ES, también se puede presentar como una manifestación visceral propia de la enfermedad (afección renal, respiratoria) sin afectación de la piel. Las consecuencias de las lesiones vasculares, las alteraciones inmunitarias y la fibrosis hística son las que dan lugar a las manifestaciones y complicaciones características de la enfermedad y su participación, en desigual magnitud en los enfermos, la responsable de la poliédrica expresión clínica de la esclerodermia 4-6.
Por tanto, en la ES, se consideran varias formas o subtipos clínicos, esclerodermia sistémica difusa en la cual se observan los cambios característicos de la piel en la región anterior del cuello y tórax (fig. 1), en la mano, se observan lesiones en garra por las retracciones de piel y tendones (fig.2), en el codo en flexus y lesiones de piel “en sal y pimienta” (fig. 3).
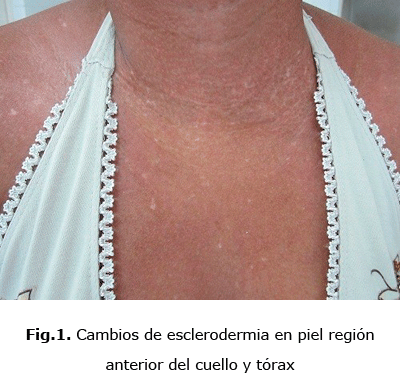
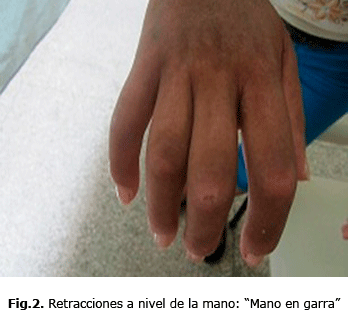
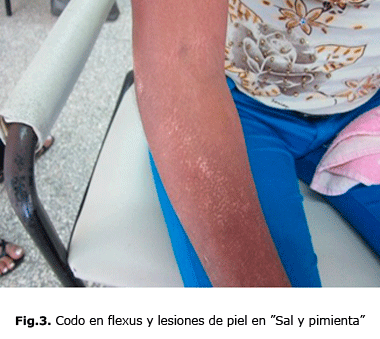
La esclerodermia sistémica limitada (ESL) cuyo sinónimo en la literatura es: síndrome de CREST (calcinosis, fenómeno de Raynaud, dismotilidad esofágica, esclerodactilia, telangiectasias), la esclerosis sistémica sin esclerodermia y las llamadas superposiciones, conforman las diferentes formas clínicas con la variabilidad en la proyección del conjunto de síntomas y signos que se presentan, según cada paciente 1-5. (tabla I).
Hasta la actualidad no existe ninguna hipótesis unificada que explique su patogenia, sin embargo, al menos anormalidades fundamentales en tres tipos de células, están íntimamente implicadas en el desarrollo de las manifestaciones clínicas y patológicas de la enfermedad. Estos tipos son: fibroblastos, células endoteliales y células del sistema inmunológico, principalmente linfocitos T y B. Las alteraciones funcionales en estas células ocasionan la característica tríada de cambios patológicos en la enfermedad, importante fibrosis cutánea y visceral, obliteración de la luz de pequeñas arterias y arteriolas, así como anormalidades en la inmunidad humoral y celular 1-3.
Tabla I. Clasificación clínica de la esclerosis sistémica
| Subtipo Clínico | Característica |
| Esclerodermia cutánea difusa | Engrosamiento cutáneo en el tronco, la cara, la extremidades proximales y dístales. |
| Esclerodermia cutánea limitada | Engrosamiento cutáneo limitado a zonas dístales al codo y la rodilla, pero también afecta el cuello y la cara. Sinónimo: síndrome de CREST (calcinosis, fenómeno de Raynaud, dismotilidad esofágica, esclerodactilia, telangiectasias). |
| Sin esclerodermia | Manifestaciones características de los órganos internos, alteraciones vasculares y serológicas, pero sin patología cutánea clínicamente detectable. |
| Superposiciones | Cualquiera de las clasificaciones previas, más lupus eritematoso sistémico, artritis reumatoidea, miopatía inflamatoria |
| Enfermedad indiferenciada del tejido conjuntivo | Fenómeno de Raynaud con manifestaciones clínicas y serológicas de ES (ulceraciones digitales, asas capilares del pliegue ungueal anormales, anticuerpos anticentrómero, edema de los dedos), pero sin engrosamiento cutáneo y sin alteraciones de los órganos internos de la ES. |
Fuente: Alarcón de Segovia 10 (2007)
Este artículo destaca algunos elementos, que según la coincidencia de diversos autores son importantes en su posible relación dentro de la compleja patogenia, con la intención de trabajar en la búsqueda de efectivas terapias para esta enfermedad incurable 4-6.
Entre los posibles agentes causales se habla de factores genéticos que contribuyen a su desarrollo. Para apoyar esta idea se observaron en varios miembros de una familia afectados con la enfermedad:
-
la alta frecuencia de trastornos autoinmunes y anticuerpos en familiares de pacientes con ES
-
las diferencias en la prevalencia
-
las manifestaciones clínicas en grupos de diferentes etnias
-
la elevada prevalencia de ciertos alelos de antígenos leucocitarios humanos (HLA)
-
la elevada prevalencia del complejo mayor de histocompatibilidad (CMH) en grupos étnicos distintos y en pacientes con diferentes subtipos clínicos de la enfermedad o diferentes tipos de autoanticuerpos 7-9.
Existe fuerte evidencia de que la producción de autoanticuerpos en la ES está determinada principalmente por factores genéticos. A pesar de que la concordancia de la enfermedad en mellizos idénticos es sólo del 4,2%, no diferente de lo que ocurre en mellizos no idénticos (5,9%), la concordancia en la presencia de anticuerpos específicos es substancialmente mayor 1. Estas observaciones indican que los factores genéticos son importantes para la producción de anticuerpos, pero no son suficientes para el desarrollo de la enfermedad, y que probablemente el factor más importante es el ambiental o de origen adquirido 10-13.
DESARROLLO
Algunos estudios sugieren que la producción de auto anticuerpos se debe a un proceso de “mimetismo molecular”. El concepto de mimetismo molecular sugiere que los anticuerpos en contra de antígenos propios se producen porque estos antígenos contienen epítopes que tienen estructura similar a la de las proteínas virales o bacterianas, entre ellos el virus del herpes, los retrovirus y el citomegalovirus humano. La hipótesis retroviral está sustentada por la existencia de homologías entre ciertas proteínas retrovirales y la proteína topoisomerasa I, la cual es el antígeno reconocido por los anticuerpos anti-Scl-70 y también por el hecho de que la expresión inducida de proteínas retrovirales en los fibroblastos normales humanos resulta en la adquisición de un fenotipo similar al de la ES con un incremento en la producción de proteínas de la matriz extracelular, además, se detectan anticuerpos contra proteínas retrovirales en el suero de los pacientes 12-15.
Algunos estudios muestran que la exposición al polvo de sílice y a ciertos metales pesados está relacionada con la etiopatogenia de la enfermedad 6, 7. Por otro lado, la exposición a disolventes orgánicos puede ser un importante factor ambiental que desencadene la enfermedad. De hecho, los individuos expuestos a cloruro de vinilo tienen un mayor riesgo de desarrollar engrosamiento de la piel, fenómeno de Raynaud, y úlceras digitales 13-16. También exposiciones a otros agentes ambientales se asocian con el desarrollo de la enfermedad, incluso ciertos plaguicidas, tintes del pelo, los llamados productos eskleros de la industria cosmética, y más recientes, los implantes de silicona, humos industriales o procedentes del petróleo 5, 6.
Una hipótesis relativamente nueva en la etiología de la ES es la del microquimerismo. Esta postula que durante el embarazo las células fetales o de origen materno atraviesan la placenta en un tránsito bidireccional. Estas células alogeneicas se injertan y permanecen en la circulación y los tejidos de la madre o del feto, debido a la compatibilidad inmunológica, se pueden activar por un evento subsiguiente y pueden iniciar una reacción de “injerto contra huésped” que se manifiesta clínicamente como ES. Las semejanzas en las manifestaciones clínicas, histopatológicas y serológicas entre la reacción “injerto contra huésped” y la ES proveen fundamento a esta hipótesis. Estas semejanzas incluyen el compromiso cutáneo, esofágico y pulmonar, así como, la producción de autoanticuerpos específicos 13-15.
Esta hipótesis es postulada inicialmente como resultado de la demostración de células fetales en la circulación de mujeres normales mucho tiempo (décadas) después del parto. Posteriormente, se identificaron ADN y células fetales en los tejidos afectados en mujeres con la enfermedad13. De acuerdo con esta hipótesis, es posible explicar la gran frecuencia de la enfermedad en mujeres.
También se encuentran células microquiméricas de origen materno en la circulación de sus descendientes, que permite explicar, la presentación de la enfermedad en hombres y en mujeres nulíparas. Aunque algunos estudios encuentran que la frecuencia de microquimerismo es similar en mujeres con ES y en mujeres sin la enfermedad, la cuantificación de estas células demuestra que su número es mucho más elevado en pacientes enfermos que en personas normales, lo cual indica que no es la mera presencia de estas células, sino su número, el factor importante en la patogenia de la enfermedad. Este estudio, demuestra que una estimulación inmunológica capaz de iniciar una reacción de linfocitos T específica para un antígeno causa, en los pacientes, diferente a los normales, una expansión muy potente de las células microquiméricas17. A pesar de estos estudios, hasta la actualidad, el papel de las células microquiméricas en la patogenia de la enfermedad no es esclarecida completamente 13-17.
Algunos elementos que tienen un papel protagónico en la etiopatogenia de la enfermedad pueden encontrarse:
-
Alteraciones de la inmunidad humoral: como la presencia de anticuerpos específicos es una de las características más comunes de ES y más del 90% de los pacientes tienen anticuerpos antinucleares; algunos son altamente específicos para la enfermedad y otros se asocian con diferentes manifestaciones clínicas 18.
Los anticuerpos anti-Scl-70 reaccionan con la ADN topoisomerasa I y se encuentran casi exclusivamente en el suero del 30% al 40% de los pacientes con la forma difusa de la enfermedad. Los anticuerpos anticentrómero están presentes entre el 80% al 90% de los pacientes con la forma limitada de la ES y sólo se encuentran en menos en el 10% de los casos con la forma difusa. Estos dos anticuerpos raramente se presentan juntos en el mismo paciente.
Otros anticuerpos menos comunes incluyen los anticuerpos anti-ARN polimerasas I y III, encontrados en pacientes con progresión rápida y compromiso visceral grave; los anticuerpos antifibrilarina, se encuentran en la forma difusa y los anti-PM-Scl en pacientes con ES que desarrollan una miopatía inflamatoria. Aunque los autoanticuerpos son comunes en la ES, no están implicados directamente en las manifestaciones clínicas de la enfermedad. Sin embargo, debido a su alta frecuencia y especificidad por ciertos subtipos clínicos su presencia es de gran ayuda para establecer el diagnóstico y pronóstico 18, 19.
-
La inflamación tisular como evento primario en la patogenia: la posibilidad de que un proceso inflamatorio crónico y persistente sea el mecanismo inicial en la patogenia de la enfermedad fue sugerida por la presencia de infiltración linfocitaria en la piel afectada de pacientes con inicio reciente de la enfermedad 19, 20. Estudios posteriores correlacionaron la intensidad de los infiltrados inflamatorios con la gravedad y la rapidez en la progresión de la enfermedad 21. Las células mononucleares presentes en los infiltrados son predominantemente linfocitos T CD4+ y expresan el antígeno DR de la clase II del CMH, que es un indicador de activación inmunológica 22-24.
La población de células inflamatorias que resultan de la expansión oligoclonal es activada, ya sea por contacto con el antígeno o por mecanismos autocrinos. Las células activadas producen citocinas y factores de crecimiento proinflamatorios y profibrosis, los cuales inician y perpetúan el proceso de fibrosis y causan las lesiones endoteliales y vasculares 25.
Numerosas observaciones confirman el estado de activación inmunológica de células T:
1) el suero de pacientes con ES contiene, aproximadamente, tres veces más IL-2R soluble que el suero normal
2) los pacientes con un curso grave y progresivo de la enfermedad tienen un número mayor de receptores que los pacientes con un curso más benigno
3) las células mononucleares del lavado broncoalveolar de los pacientes con compromiso pulmonar se encuentran activadas
4) niveles elevados de IL-2 en el suero y su producción excesiva por células T en cultivo, en pacientes con ES. Sin embargo, no está claro si las alteraciones en las proporciones de los subtipos de células T y de las citocinas son un efecto secundario de la actividad de la enfermedad o representan un evento fundamental en su patogenia 27.
-
Papel del TGF-β, y proteínas Smad: actualmente, no se sabe si la producción exagerada de tejido conectivo por los fibroblastos con ES representa una regulación anormal de un proceso fisiológico en respuesta al agente etiológico desconocido, o si el evento principal es una alteración de la regulación de la expresión de los genes de las proteínas relevantes de la matriz. En la ES se encuentran numerosas alteraciones en la expresión de las citocinas y los factores de crecimiento, que producen serios efectos en la síntesis de colágeno por los fibroblastos, en las funciones de células endoteliales y en las respuestas de las células T 27-30.
Luego, una serie de reacciones en cadena ocurre a través de las proteínas de la familia Smad. La Smad 2 ó la Smad 3 son fosforiladas por el complejo de los dos receptores del TGF-β. Una vez fosforiladas, la Smad 2 o la Smad 3 forman un complejo con la proteína citoplasmática Smad 4, la cual permite la translocación del complejo Smad al interior del núcleo. La Smad 7 es una proteína inhibitoria que puede prevenir la fosforilación de Smad 2 o Smad 3. Parece que el IFNγ y el TNF-α, dos potentes citocinas inhibitorias de la síntesis de colágeno, estimulan el incremento de la expresión de Smad 7, lo que ocasiona una reducción de la expresión del gen de colágeno inducido por el TGF-β 31-36.
-
Fibrosis tisular y vascular: las manifestaciones clínicas más prominentes de la ES se deben a la acumulación exagerada de colágeno y otros componentes del tejido conectivo en los órganos afectados, debida a la producción excesiva de colágeno por los fibroblastos. Cualquiera que sea el suceso etiológico, son cruciales las alteraciones consecuentes en el fenotipo biosintético de los fibroblastos en la patogenia de la enfermedad 33-36.
CONCLUSIONES
Mecanismos etiopatogénicos basados en anormalidades moleculares, pueden ser correctas en la ES. Su espectro clínico heterogéneo responde a una etiopatogenia distinta en cada individuo, así como a eventos complejos que pueden ocurrir en fases iniciales de la enfermedad y no a una patogenia única. Hipótesis en cuanto a factores genéticos, hereditarios, ambientales, de origen infeccioso, forman parte de los elementos detonadores del disturbio inmunológico y las reacciones individuales en el huésped susceptible.
REFERENCIAS BIBLIOGRÁFICAS
1. Daoussis D, Antonopoulos I, Liossis SN, Yiannopoulos G, Andonopoulos AP. Treatment of Systemic Sclerosis-Associated Calcinosis: A Case Report of Rituximab-Induced Regression of CREST-Related Calcinosis and Review of the Literature. Semin Arthritis Rheum.2012 [citado 25 abr 2015]; 41(6):822-829. Disponible en: http://www.sciencedirect.com/science/article/pii/S004901721100357X
2. Chakravarty E. Pre-disease pregnancy complications and systemic sclerosis: pathogenic or pre- clinical? .Arthritis Res Ther. 2012[citado 25 abr 2014]; 14(1):1-2. Disponible en: http://www.arthritis-research.com/content/pdf/ar3686.pdf
3. Milanetti F, Bucha J, Testori A, Burt RK. Autologous hematopoietic stem cell transplantation for systemic sclerosis. Curr Stem Cell Res Ther.2011 [citado 7 dic 2015]; 6(1):16-28.Disponible en: http://www.eurekaselect.com/87399/article/autologous-hematopoietic-stem-cell-transplantation-systemic-sclerosis
4. Mouthon L, Bérezné A, Bussone G, Noël LH, Villiger PM, Guillevin L. Scleroderma renal crisis: a rare but severe complication of systemic sclerosis. Clin Rev Allerg Immunol. 2011 [citado 25 abr 2014]; 40(2):84-91.Disponible en: http://link.springer.com/article/10.1007%2Fs12016-009-8191-5
5. Becker MO, Müller-Ladner U, Riemekasten G. Towards an implementation of guidelines for the therapy of systemic sclerosis (scleroderma): between desire and reality. Z Rheumatol. 2010 [citado 20 abr 2015]; 69(4):310-317.Disponible en: http://europepmc.org/abstract/MED/20490516
6. Dimitroulas T, Giannakoulas G, Karvounis H, Settas L, Kitas GD. Systemic sclerosis-related pulmonary hypertension: unique characteristics and future treatment targets. Curr Pharm Des. 2012 [citado 25 abr 2014]; 18 (11):1457-1464.Disponible en: http://www.ingentaconnect.com/content/ben/cpd/2012/00000018/00000011/art00003
7. Sfikakis PP, Papamichael C, Stamatelopoulos KS, Tousoulis D, Fragiadaki KG, Katsichti P, et al. Improvement of vascular endothelial function using the oral endothelin receptor antagonist bosentan in patients with systemic sclerosis. Arthritis Rheum.2007 [citado 18 mar 2015]; 56(6):1985-1993.Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/art.22634/full
8. Walker UA, Tyndall A, Czirja kL, Denton C, Farge-Bancel D, Kowal-Bielecka O, et al. Clinical risk assessment of organ manifestations in systemic sclerosis: a report from the EULAR Scleroderma Trials and Research group database. Ann Rheum Dis. 2007[citado 18 mar 2015]; 66(6):754–763.Disponible en: http://ard.bmj.com/content/66/6/754.abstract
9. Simeón CP, Fonollosa V, Vilardell M. Epidemiología y clasificación de la esclerosis sistémica (esclerodermia).Barcelona. Marge Médica Books; 2009.
10. de Segovia A. Tratado Hispanoamericano de Reumatología: Vol. II. Bogotá: Schering Plough; 2007.
11. Van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et.al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013[citado 25 abr 2014]; 65(11):2737-2747.Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/art.38098/abstract
12. Johnson SR, Brode SK, Mielniczuk LM, Granton JT. Dual therapy in IPAH and SSc-PAH.A qualitative systematic review. Respir Med. 2012[citado 25 abr 2015]; 106(5):730-739. Disponible en: http://www.resmedjournal.com/article/S0954-6111%2812%2900028-5/pdf
13. García de la Peña Lefebvrea P .Aspectos clínicos novedosos en la esclerodermia. Reumatol Clin.2008 [citado 22 may 2015]; 4(1):45-49. Disponible en: http://www.reumatologiaclinica.org/es/aspectos-clinicos-novedosos-esclerodermia/articulo/S1699258X08761400/
14. Walker KM, Pope J. Treatment of Systemic Sclerosis Complications: What to Use When First-Line Treatment Fails-A Consensus of Systemic Sclerosis Experts. Semin Arthritis Rheum.2012 [citado 25 abr 2014]; 42(1):42-55. Disponible en: http://www.semarthritisrheumatism.com/article/S0049-0172%2812%2900017-0/pdf
15. John Varga MD. Systemic Sclerosis: An Update. Bull NYU Hosp Jt Dis. 2008[citado 7 dic 2015]; 66(3):198-202.Disponible en: http://www.hjdbulletin.org/Archives/view/27
16. Yamada D, Asano Y, Takahashi T, Masui Y, Aozasa N, Akamata K, et al. Clinical significance of serum decoy receptor 3 levels in patients with systemic sclerosis. Eur J Dermatol.2012 [citado 25 abr 2014]; 22(3):351-357.Disponible en: http://www.jle.com/fr/revues/ejd/e-docs/clinical_significance_of_serum_decoy_receptor_3_levels_in_patients_with_systemic_sclerosis_293022/article.phtml
17. Li N, Elashoff RM, Li G, Tseng CH. Joint analysis of bivariate longitudinal ordinal outcomes and competing risks survival times with nonparametric distributions for random effects. Stat Med. 2012[citado 25 abr 2014]; 31(16):1707-1721.Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/sim.4507/full
18. Van Laar JM, Stolk J, Tyndall A. Scleroderma lung.Pathogenesis, evaluation and current therapy.Drugs.2007 [citado 25 abr 2014]; 67(7):985-996.Disponible en: http://link.springer.com/article/10.2165/00003495-200767070-00004#page-1
19. Brito-Zerón P, Sisó-Almirall A, Bové-Boa-da A, Ramos-Casals M. Diagnóstico y tratamiento de la enfermedad vascular periférica en la esclerosis sistémica (esclerodermia).En: Avances en enfermedades autoinmunes sistemática: Avances en esclerosis sistémica (esclerodermia).Barcelona: Marge Médica Books; 2009. [citado 7 dic 2015];87-102.Disponible en: https://books.google.es/books?id=TifPf9J_gAYC&pg=PA1
20. Yi T, Song SU. Immunomodulatory properties of mesenchymal stem cells and their therapeutic applications. Arch Pharm Res. 2012 [citado 25 abr 2015]; 35(2):213-221. Disponible en: http://link.springer.com/article/10.1007%2Fs12272-012-0202-z
21. Avouac J, Walker U, Tyndall A, Kahan A, Matucci-Cerinic M, Allanore Y. Characteristics of joint involvement and relationships with systemic inflammation in systemic sclerosis: results from the EULAR Scleroderma Trial and Research. J Rheumatol.2010 [citado 25 abr 2014]; 37(7):1488-1501. Disponible en: http://www.jrheum.org/content/37/7/1488
22. Cuomo G, Abignano G, Ruocco L, Vettori S, Valentini G. Hypocomplementemia in systemic sclerosis. Reumatismo. 2008[citado 25 abr 2014]; 60(4):268-273. Disponible en: http://www.reumatismo.org/index.php/reuma/article/view/reumatismo.2008.268
23. Hinchcliff M, Varga J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am Fam Physician. 2008[citado 3 dic 2015]; 78(8):961-968.Disponible en: http://www.aafp.org/afp/2008/1015/p961.html
24. Mouthon L, Rannou F, Bérezné A, Pagnoux C, Arène JP, Foïs E, et al. Development and validation of a scale for mouth handicap in systemic sclerosis: the Mouth Handicap in System Sclerosis scale. Ann Rheum Dis.2007 [citado 3 dic 2015]; 66(12):1651-1655. Disponible en: http://ard.bmj.com/content/66/12/1651.abstract
25. Arnaud L, Huart A, Plaisier E, Francois H, Mougenot B, and Tiev K, et al. ANCA-related crescentic glomerulonephritis in systemic sclerosis: revisiting the “normotensive scleroderma renal crisis”. Clin Nephrol.2007 [citado 25 abr 2014]; 68(3):165-170.Disponible en: http://europepmc.org/abstract/med/17915619
26. Sfikakis PP, Papamichael C, Stamatelopoulos KS, Tousoulis D, Fragiadaki KG, Katsichti P, et al. Improvement of vascular endothelial function using the oral endothelin receptor antagonist bosentan in patients with systemic sclerosis. Arthritis Rheum. 2007[citado 25 abr 2014]; 56(6):1985-1993.Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/art.22634/abstract
27. Zaima C, Kanai M, Ishikawa S, Kawaguchi Y, Masui T, Mori Y, et al. A case of progressive digital ischemia after early withdrawal of gemcitabine and S-1 in a patient with systemic sclerosis. Jpn J Clin Oncol. 2011[citado 4 dic 2015]; 41(6):803-806.Disponible en: http://jjco.oxfordjournals.org/content/41/6/803.short
28. Tashkin DP, Elashoff R, Clements PJ, Roth MD, Furst DE, Silver RM, et al. Effects of 1-year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med.2007 [citado 4 dic 2015]; 176(10):1026-1034.Disponible en: http://www.atsjournals.org/doi/abs/10.1164/rccm.200702-326OC#.Vl2_NfssfaF
29. Paone C, Chiarolanza I, Cuomo G, Ruocco L, Vettori S, Menegozzo M, et al. Twelve-month azathioprine as maintenance therapy in early diffuse systemic sclerosis patients treated for 1-year with low dose cyclophosphamide pulse therapy. Clin Exp Rheumatol. 2007[citado 4 dic 2015]; 25(4):613-616.Disponible en: http://www.clinexprheumatol.org/article.asp?a=3126
30. Distler JH, Jüngel A, Huber LC, Schulze-Horsel U, Zwerina J, Gay RE, et al. Imatinib mesylate reduces production of extracellular matrix and prevents development of experimental dermal fibrosis. Arthritis Rheum.2007 [citado 4 dic 2015]; 56(1):311-322.Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/art.22314/abstract
31. Meune C, Avouac J, Wahbi K, Cabanes L, Wipff J, Mouthon L, et al .Cardiac Involvement in Systemic Sclerosis Assessed byTissue-Doppler Echocardiography During Routine Care .A Controlled Study of 100 Consecutive Patient. Arthritis Rheum. 2008[citado 7 dic 2015]; 58(6): 1803–1809.Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/art.23463/abstract
32. Fertig N, Domsic RT, Rodríguez- Reyna T, Kuwuana M, Lucas M, Medsger TA, et al. Anti–U11/U12 RNP Antibodies in Systemic Sclerosis: A New Serologic Marker Associated With Pulmonary Fibrosis. Arthritis Rheum.2009 [citado 7 dic 2015]; 61(7):958–965.Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/art.24586/abstract
33. Hershcovici T, Jha L K, Johnson T, Gerson L, and Stave C, Malo J.Systematic review: the relationship between interstitial lung diseases and gastro-oesophageal reflux disease. Aliment Pharmacol Ther. 2011[citado 8 dic 2015]; 29(68):85-92.Disponible en: http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2036.2011.04870.x/full
34. Valentini G, Romano M, Naclerio C, Bisogni R, Lamberti A, Turco MC, et al. Increased expression of CD40 ligand in activated CD4+ T lymphocytes of systemic sclerosis patients. J Autoimmun.2000 [citado 8 dic 2015]; 15(1):61-66.Disponible en: http://www.sciencedirect.com/science/article/pii/S0896841100903871
35. Anaya JM, Shoenfeld Y, Correa PA, Carrasco García M, Cervera R. Autoinmunidad y Enfermedad Autoinmune. Medellin: Corporación para Investigaciones Biológicas; 2005[citado 8 dic 2015].Disponible en: http://es.scribd.com/doc/39104877/Autoinmunidad-y-Enfermedad-Autoinmune-1era-Ed-2005#scribd
36. Remedios Batista SE, Velázquez Grass A, Del Campo Avilés E, Torres Pérez L, Fernández Portelles A. Ciclofosfamida en el tratamiento de la esclerosis sistémica. CCM. 2015 [citado 28 ene 2016]; 19(4): 706-717. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1560-43812015000400010&lng=es
Recibido: 7 de mayo de 2015
Aprobado: 30 de noviembre de 2015
Dra. Susel E. Remedios Batista. Hospital Clínico Quirúrgico Carlos Font. Banes. Holguín. Cuba.
Correo electrónico: remesusy@banes.hlg.sld.cu

