










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Endocrinología
versión On-line ISSN 1561-2953
Rev Cubana Endocrinol v.13 n.3 Ciudad de la Habana sep.-dic. 2002
Presentación de casos
Hospital Pediátrico Docente "William Soler"
Hospital Clinicoquirúrgico "Hermanos Ameijeiras"
Disgenesia gonadal mixta con fórmula cromosómica 45,X/46,X, (mar). Presentación de una paciente
Dr. Pedro González Fernández,1 Lic. Marlene Quesada Dorta,2 Dra. Raquel Cabrera Panizo3 y Lic. Daysi Bello Álvarez4
Resumen
Se presentó una paciente con estigmas turnerianos y cariotipo 45,X/46,XX con diagnóstico inicial de síndrome de Turner a la que se le realizó clitoridectomía por hipertrofia del clítoris a los 8 meses de edad. Se reevaluó a los 6 años de edad y se le realizó cariotipo con técnicas de bandas G (GTG) con fórmula cromosómica de 45,X/46,X, (cromosoma marcador -mar); dicho marcador dio la impresión de una deleción del cromosoma X desde Xq13 ®Xq ter y Xp22 ®Xp ter. Se completó dicho estudio con técnica molecular de reacción en cadena de la polimerasa (PCR) y se identificó el gen SRY en el cromosoma marcador. Se realizó intervención quirúrgica por mínimo acceso y se comprobó ausencia de útero así como trompa en el lado derecho con ausencia de gónada y en el lado opuesto, testículo rudimentario; se planteó el diagnóstico de disgenesia gonadal mixta.
DeCS: DISGENECIA GONADAL MIXTA; SINDROME DE TURNER; SEUDOHERMAFRODITISMO; MASCULINO; FEMENINO.
El desarrollo sexual se inicia en el momento de la concepción al establecerse el sexo cromosómico. A la quinta semana de vida intrauterina comienza la formación de la gónada bipotencial la que, en dependencia del sexo cromosómico, dará lugar a una gónada masculina o femenina. Cualquier causa que interfiera en estos procesos en las primeras semanas de vida fetal dará lugar a los trastornos de la diferenciación sexual.1
Aunque la atención de estos pacientes es difícil y requiere casi siempre de medios y técnicas complejas, los hallazgos al realizar el examen físico siguen ocupando un importante lugar en el correcto análisis diagnóstico.
El diagnóstico temprano y correcto es importante no sólo por las implicaciones familiares, cosméticas y sociales sino porque en algunas entidades como la disgenesia gonadal mixta (DGM) la gónada disgenética pudiera tener una alta potencialidad de malignización que, de no tomarse la conducta adecuada, originaría consecuencias fatales.2
Tomando en consideración todo lo anterior así como la infrecuencia de la fórmula cromosómica, presentamos el caso de una paciente con estigmas turnerianos y cariotipo 45,X/46,X, (mar) en quien los hallazgos al realizar el examen físico así como la técnica molecular para identificar material cromosómico del Y fueron decisivos para el diagnóstico de DGM.
Presentación de la paciente
Paciente con fenotipo femenino, procedente de Villa Clara, con antecedentes de parto normal, peso de 6 lib y la talla fue de 48 cm al nacer, al efectuar el examen físico se observa cuello corto, ancho y discretamente alado; tórax en escudo con teletelia e hipertrofia del clítoris. Se realizó cromatina oral 0 % y cariotipo 45,X/46,XX y se plantea diagnóstico de síndrome de Turner (ST), se le practicó clitoridectomía a los 8 meses de edad.
A los 6 años de edad acude al Servicio de Endocrinología de este hospital para continuar seguimiento e iniciar tratamiento hormonal. En ese momento se encuentra al examen físico: cuello corto, ancho y discretamente alado; tórax en escudo con teletelia y pezones invertidos; ligera rizomelia, así como baja talla (106 cm) y coeficiente intelectual de 113 (figs. 1 y 2).

FIG.1. Vista de perfil de la paciente.

FIG.2. Vista de espalda de la paciente.
Dados los antecedentes en la anamnesis y el examen físico se decide realizar nuevamente cromatina sexual en mucosa oral cuyo resultado es de 0 %, así como cariotipo con técnicas de bandas G (GTG) que arroja una fórmula cromosómica de 45,X/46,X, (mar)? en 40 metafases analizadas, dicho marcador da la impresión de una deleción del cromosoma X desde Xq13®Xq ter y Xp22®Xp ter (p: brazo corto. q : brazo largo. ter: Terminal. International System Chromosome Nomenclature) (figs. 3 y 4).
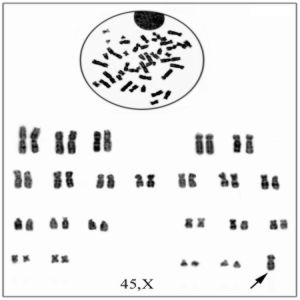
FIG.3. Se observa la línea celular con fórmula cromosómica 45, X. El cromosoma se señala con una flecha.

FIG. 4. Se observa la línea celular con fórmula cromosómica 46,X, (mar). El cromosoma mar se señala con una flecha.
Se decide completar dicho estudio con la técnica molecular de reacción en cadena de la polimerasa (PCR), se identifica el gen SRY en el cromosoma marcador. Se plantea diagnóstico de disgenesia gonadal mixta, se realiza intervención quirúrgica por mínimo acceso y se comprueba la ausencia de útero, presencia de trompa en el lado derecho con ausencia de gónada, y en el lado contralateral, testículo rudimentario.
Discusión
Los trastornos de la diferenciación sexual constituyen un grupo complejo de entidades y síndromes. Una clasificación muy socorrida es la de agrupar a estos pacientes en pseudohermafroditismo masculino (PM), pseudohermafroditismo femenino (PF) y hermafroditismo verdadero (HV) según la presencia de una gónada masculina, femenina o la presencia de ambas estructuras (ovotestes), respectivamente.1
La DGM es la forma más frecuente de PM y se caracteriza por un testículo disgenético unilateral y una gónada contralateral en cordón o bandeleta (streak gonad) así como defectos en la virilización y persistencia de los conductos müllerianos. Aunque en términos rigurosos no debería ser clasificado como PM, puesto que no hay testículos normales, su inclusión en este grupo ayuda a esclarecer el proceso del desarrollo testicular normal y permite ubicarnos al analizar el diagnóstico de un paciente con trastornos de la diferenciación sexual.2
Se ha sugerido una relación estrecha entre DGM y ST porque el cariotipo 45,X/46,XY que es típico de la DGM, puede observarse en pacientes con fenotipo ST 3 y los hallazgos fenotípicos del ST pueden verse en los pacientes con DGM.4,5
A esta paciente se le diagnosticó inicialmente un ST, tomando en consideración los estigmas turnerianos, pero si bien es cierto que estos hallazgos podrían sugerir ese diagnóstico, la presencia de virilización en esta paciente lo descartaría. La ocurrencia de estigmas turnerianos en pacientes con anomalías del cromosoma Y está relacionado con la coexistencia de una línea celular 45, X.6
En una serie de 82 pacientes7 se presentaba la baja talla como único estigma turneriano, 13 de ellos tenía un mosaico con una línea celular con un cromosoma Y isodicéntrico. Sin embargo, Giltay, et al.8 informaron de un paciente similar en quien se encontró un gonadoblastoma.
El brazo corto del cromosoma Y contiene el gen para iniciar la diferenciación testicular conocida como región determinante del sexo del cromosoma Y (SRY). Una anomalía en el brazo corto del cromosoma Y daría lugar a la presencia de estrías gonadales o disgenesia testicular; ya que no se produciría el organizador testicular y los genitales internos y externos serían ambiguos, pues las secreciones del factor inhibidor de Müller y de testosterona estarían alterados por los testículos anormales, es decir, con la pérdida del brazo corto del cromosoma Y no se forman los testículos y por eso se desarrolla un fenotipo femenino. La pérdida parcial del cromosoma X en algunas células (X0/XY) produciría DGM, la que daría lugar a una de estas variantes: un testículo inmaduro de un lado y una gónada rudimentaria del otro; agenesia gonadal unilateral; gónadas hipoplásicas bilaterales intraabdominales con elementos rudimentarios testiculares en una de ellas o gonadoblastoma unilateral o bilateral.9
Por la alta posibilidad de malignización de la gónada disgenética en estas entidades es que se hace necesario el correcto y temprano diagnóstico, para evitar la posibilidad de que se transformen en gonadoblastoma.10,11
Lieppe,12 en 205 pacientes con diagnóstico de síndrome de Turner encontró uno solo con fórmula cromosómica 45,X/46,XX (mar) del cromosoma Y en el que al extirpársele la gónada se diagnosticó un gonadoblastoma. Un paciente con fenotipo turneriano y fórmula cromosómica similar a la de nuestra paciente fue informado por Hashimoto, et al.,5 pero no hayamos referencia de un caso similar publicado en nuestro país.
Aunque la frecuencia de gonadoblastoma es baja en pacientes con fenotipo y cariotipo de ST, se recomienda la búsqueda de material del cromosoma Y en aquellos con signos de virilización.13 Por otra parte, el gen responsable del gonadoblastoma (GBY) se localiza cerca del centrómero y no en la parte distal del brazo corto del cromosoma Y, donde se halla el gen SRY, por lo cual Page14 plantea que en ausencia de centrómero, la presencia o no del gen SRY tiene escaso valor como elemento de riesgo para el gonadoblastoma, por lo que la técnica molecular de hibridización in situ fluorescente (FISH) con prueba centromérica específica sería lo aconsejable para aseverar el riesgo de gonadoblastoma. En nuestro medio nosotros no disponemos de esa técnica por lo que no fue posible realizarla a esta paciente.
El PM es la indicación más frecuente para la laparoscopia como procedimiento terapéutico. Denes, et al.15 en 29 pacientes con trastornos de la diferenciación sexual y edad entre 6 meses y 46 años, hallaron 20 sujetos con PM de los cuales 16 eran DGM.
Como era de esperar en esta paciente, en el proceder laparoscópico se encontraron esbozos de estructuras müllerianas como resultado de la deficiencia de la hormona inhibidora de las estructuras müllerianas secretada por las células de Sertoli, así como ausencia de gónada en el lado derecho y en el izquierdo, un testículo rudimentario.2
Nuestra paciente tenía una fórmula cromosómica infrecuente así como estigmas turnerianos y signos de virilización. Si bien es cierto que el diagnóstico de los trastornos de la diferenciación sexual constituye un tópico complejo por los distintos aspectos (cromosómicos y hormonales, entre otros) involucrados en su patogenia y los indefectibles y sofisticados medios diagnósticos, los hallazgos clínicos continúan ocupando un lugar clave para el correcto diagnóstico de estos pacientes.
Summary
A patient with Turner stigmas and karyotype 45,X/46,XX with initial diagnosis of Turner's syndrome is presented. Clitoridectomy was performed due to hypertrophy of the clitoris when she was 8. She was reevaluated at 6 and karyotype was made by using G bands techniques (GTG) with chromosomic formula of 45,X/46,X, (marker chromosome -mar); such marker gave the impression of a deletion of chromosome X from Xq13 ®Xq ter and Xp22 ®Xp ter. This study was completed with the molecular technique of polymerase chain reaction (PCR) and the SRY gene was identified in the marker chromosome. The patient underwent minimum access surgery and it was proved the absence of uterus and tube on the right side with no gonad and a rudimentary testis on the opposite side. Mixed gonodal dysgenesis was diagnosed.
Subject headings: DYSGENESIS, MIXED; TURNER'S SYNDROME PSEUDOHERMAPHRODITISM; MALE; FEMALE.
Referencias bibliográficas
- Grumbach MM, Conte FA. Disorders of sex differentiation. En: Wilson JD, Foster DW, Kronenberg HM, Larsen PR, Ed. Williams Textbook of Endocrinology. 9th ed. Philadelphia: Saunders, 1998.P. 1303-425.
- Jubiz W. Diferenciación sexual: hermafroditismo. En: Ávila Valdivieso JJ, Ed. Endocrinología Clínica. México DF: Editorial El Manual Moderno; 1996: 481-502.
- Yamakita N, Yasuda K, Mori H, Kuriyama M, Kumamoto Y, Miura K. A case of mixed gonadal dysgenesis (MGD), with a review of MGD patients reported in Japan. Jpn J Med 1989;28:744-52.
- Kofman S, Pérez-Palacios G, Medina M, Escobar N, García M, Ruz L, et al. Clinical and endocrine spectrum in patients with the 45,X/46,XY karyotype. Human Genet 1981;58:773-6.
- Hashimoto H, Maruyama H, Koshida R, Okuda N, Murayama K, Katsumi T, et al. Presence of Turner stigmata in a case of dysgenetic male pseudohermaphroditism with 45,X/46,X+ mar karyotype. Arch Dis Child 1997;76:268-71.
- Hsu LYF. Phenotype karyotype correlation of Y chromosome aneuploidy with emphasis on structural aberrations in post-natally diagnosed cases. Am J Med Genet 1994:53;108-14.
- Tuck-Muller CM, Chen H, Martinez JE, Shen CC, Li S, Kusyk C, et al. Isodicentric Y chromosome cytogenetic, molecular and clinical studies and review of the literature. Hum Genet 1995:96;119-29.
- Giltay JC, Ausens MGEM, Van Seumeren I, Zewald RA, Sinke RJ, Fass B, et al. Short stature as the only presenting feature in a patient with an isodicentric (Y) (q11,23) and gonadoblastoma. A clinical and molecular cytogenetic study. Eur J Pediatr 2001;160:154-8.
- Hughes IA, Coleman N, Faisal Ahmed S, Ng K-L, Cheng A, Lim HN, et al. Sexual dismorphism in the neonatal gonad. Acta Paediatr 1999 (Suppl);428:23-30.
- Lau YFC. Sex chromosome genetics´99. Gonadoblastoma, testicular and prostate cancers, and the TSPY gene. Am J Hum Genet 1999;64:921-7.
- Abeliovich D, Yehuda O, Ben-Neriah S, Or A. Loss of Y chromosome: an age-related event or a cytogenetic marker of a malignant clone? Cancer Genet Cytogenet 1994;76:70-1.
- Lieppe BM. Turner Syndrome. En: Sperling MA, ed. Pediatric Endocrinology. Chicago: WB Saunders Co. 1996.P. 387-421.
- Hughes IA. The masculinized female and investigation of abnormal sexual development. Baillière's Clin Endocrinol Metab 1998;12:157-71.
- Page DC. Y chromosome sequences in Turner's syndrome and risk of gonadoblastoma or virilization. Lancet 1994;343:240-5.
- Dénes FT, Mendonça BB, Arap S. Laparoscopic management of intersexual states. Urol Clin North Amer 2001;28:31-42.
Recibido: 18 de octubre de 2002. Aprobado: 19 de diciembre de 2002.
Dr. Pedro González Fernández. Calle 39 No. 873 entre Ave. 26 y 24, Nuevo Vedado, Plaza, Ciudad de La Habana, Cuba.
1 Especialista de II Grado en Endocrinología. Profesor Asistente. Hospital Pediátrico Docente "William Soler."
2 Licenciada en Biología. Profesora Asistente. Investigadora Agregada. Laboratorio de Genética y Biología Molecular. Hospital Clinicoquirúrgico "Hermanos Ameijeiras."
3 Especialista de I Grado en Pediatría. Servicio de Endocrinología del Hospital Pediátrico Docente "William Soler."
4 Licenciada en Biología. Laboratorio de Genética y Biología Molecular. Hospital Clinicoquirúrgico "Hermanos Ameijeiras".

