










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Endocrinología
versión On-line ISSN 1561-2953
Rev Cubana Endocrinol v.17 n.1 Ciudad de la Habana ene.-abr. 2006
Instituto Nacional de Endocrinología
Presentación de caso
Déficit aislado de GH tipo familiar: informe sobre el primer paciente en Cuba
Dra. Tania Espinosa Reyes,1 Dr. Rodrigo Espinoza Iturri,2 Dra. Tamara Fernández Teruel,1 Dra. Cecilia Pérez Gesen,3 Dra. Teresa Montesinos Estévez4 y Dr. Francisco Carvajal Martínez5
Resumen
El déficit de hormona de crecimiento (GH) aislado es una afección de naturaleza congénita o adquirida que se caracteriza por la ausencia parcial o total de hormona de crecimiento detectable en plasma o suero. Se presenta un paciente de 11 años, blanco, masculino, quien asiste a consulta por retraso marcado del crecimiento y desarrollo, con talla/edad correspondiente a 4,4 años y cifras de GH en pruebas farmacológicas que no superan los 10 ng/mL. El resto del estudio hormonal está dentro de los parámetros normales. Se comprueba en la madre baja talla y el estudio hormonal muestra similares resultados. La búsqueda de individuos de baja talla en la familia muestra iguales características en abuela y bisabuela maternas. Se diagnostica como un déficit aislado de GH de tipo familiar, tipo II, con patrón de herencia autosómica dominante. Se indica tratamiento con hormona de crecimiento biosintética con resultados satisfactorios. Se concluye que el estudio de la genealogía de un caso índice permite dilucidar la presencia de otros sujetos afectos y establecer los patrones de transmisión de la enfermedad genética.
Palabras clave: Déficit aislado de GH, déficit familiar.
La hormona de crecimiento (GH) aparentemente no es esencial en el crecimiento fetal. Los monos hipofisectomizados, al igual que los recién nacidos humanos con agenesia de hipófisis, tienen talla relativamente normal al nacer. Sin embargo, en la etapa posnatal constituye la principal reguladora del crecimiento somático. Esta acción la ejerce indirectamente a través de la inducción de la síntesis de otra hormona: el factor de crecimiento similar a la insulina-1 (IGF-1), cuya acción principal es estimular la síntesis de ácido desoxirribonucleico (DNA) e inducir multiplicación celular. La GH circula unida a una proteína transportadora específica (GH-bP), que tiene una secuencia aminoacídica similar a la porción extramembranosa de la GH en los tejidos, por lo que además de transportarla regula su acción ( Allen D. Endocrinology update for pediatricians: growth issues and growth hormone. Presented at the Annual Meeting of the American Academy of Pediatrics; October 29, 2000; Chicago).
El déficit de GH aislado o asociado a otras deficiencias hormonales es una afección de naturaleza congénita o adquirida caracterizada por la ausencia parcial o total de hormona de crecimiento detectable en plasma o suero. Se presenta más frecuente en varones que en niñas, con una relación 4:1.
El gen de la hormona de crecimiento humana ha sido identificado, aislado y caracterizado en el brazo largo del cromosoma 17, y se encuentra a su mismo nivel todo un complejo de genes relacionados: GH1 y GH2.2,3 La figura 1 muestra la localización y organización del cluster de hGH.
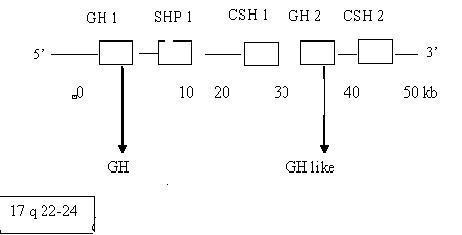
FIG. Localización y organización del cluster de hGH.
El déficit de GH constituye un síndrome en el que se presenta una combinación de anomalías auxológicas, clínicas, bioquímicas y metabólicas causadas por una secreción anormal de GH, con reducción en la generación de las hormonas y de los factores de crecimiento que dependen de ella.4 Se debe sospechar en niños con retraso de talla proporcionada, velocidad de crecimiento inferior a 4 cm al año, índice peso/talla normal o alto, adiposidad troncular, especialmente sobre pectorales o abdomen. Aspecto armónico, grácil e infantil, facie característica de muñeca sólo si el déficit se presenta desde los primeros meses de vida: frente amplia, abombada, macizo facial poco desarrollado, nariz cóncava, mejillas redondeadas, hipoplasia maxilar, piel fina y pálida y voz de tono alto por inmadurez del cartílago cricoides. Pueden tener defectos de la línea media facial tales como labio leporino, incisivo único, úvula bífida, entre otros.
El desarrollo intelectual es habitualmente normal, salvo en los casos que han presentado hipoglucemias graves en edades tempranas. La pubertad está siempre retrasada, aún en el déficit aislado de GH.5-7
En el adulto los síntomas y signos más comunes son: disminución de la masa corporal no grasa, incremento de la adiposidad abdominal, disminución de la fuerza, la vitalidad, la tolerancia al ejercicio, predisposición al desarrollo de arterosclerosis prematura, depresión y aislamiento.8 A pesar de lo controversial del tratamiento con GH en el adulto, se describe una recuperación de la masa corporal no grasa, aumento del contenido mineral óseo, incremento de la fuerza muscular y de la capacidad de ejercicio, todo lo cual mejora la calidad de vida de estos pacientes.9
Se conoce que existen diferentes variantes etiológicas genéticas que, al afectar el eje hipotálamo-hipofisario-periférico, pueden alterar la capacidad de acción o de producción de hormona de crecimiento. La mayoría tiene un patrón de herencia autosómico recesivo, excepto el déficit aislado de GH tipo 2, cuyo modo de herencia es autosómico dominante. La severidad clínica es variable entre familias y se conoce que responden bien al tratamiento con GH, como se representa a continuación:
| Déficit aislado de GH | Herencia |
| IA | AR |
| IB | AR |
| II | AD |
| III | Ligada al X |
| GH bioinactiva | AR? |
| Mutación del receptor GHRH | AR |
AR: Autosómico recesivo.
AD: Autosómico dominante.
GHRH: Hormona liberadora de hormona de crecimiento.
Presentación de caso
Paciente D.I.M. masculino, raza blanca, 11 años, procedente de área rural, producto de gestación a término de 38 semanas, parto distócico, cesárea por presentación pelviana, peso y talla normales al nacer.
Motivo de consulta: Baja talla y dificultades en el aprendizaje en los primeros años escolares, cuadro primeramente interpretado como posible hipotiroidismo secundario vs panhipopituitarismo, motivo por el cual fue remitido a nuestro centro de investigación. Durante la confección de la historia clínica, conjuntamente con los hallazgos clínicos del paciente y la disminución marcada de la velocidad de crecimiento anual, llamó la atención la talla de la madre y el tono agudo y atiplado de su voz, lo que llevó a realizar el estudio en ambos padres, con la referencia de que otros miembros de la familia materna presentaban similares características fenotípicas.
Examen físico: Apariencia armónica, voz atiplada. Tiroides: grado 0. Paratiroides: maniobra de Chvostek negativa. Adrenales: ausencia de vello sexual. Gónadas: genitales externos de aspecto masculino, testes descendidos, 2 mL de volumen. Pene: Tanner 1.
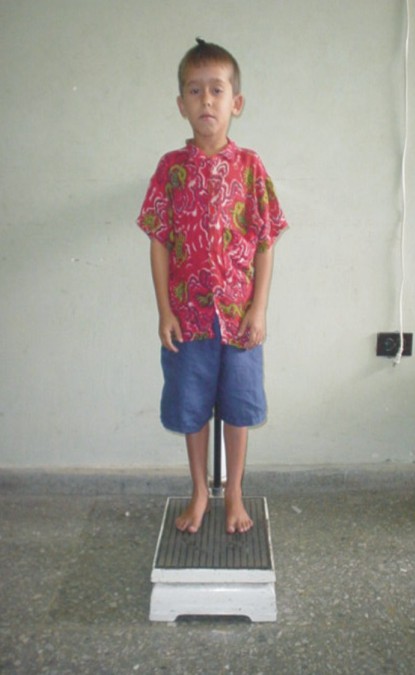
FIG. 2. Paciente con déficit de hormona de crecimiento.
Auxología: Peso: 14,5 kg. Talla: 104 cm. PC: 50 cm. PT: 49 cm. PA: 46 cm. Brazada: 104 cm. DBA: 23 cm. DBT: 20 cm. SS: 55 cm. SI: 52 cm. Peso/talla: < 3 p. Peso/edad: << 3 p (correspondiente a 2,4 años). Talla/edad: << 3 p (correspondiente a 4,4 años). Fondo de ojo: normal. Talla del padre: 165 cm. Talla de la madre: 132,2 cm. Talla esperada: 154 cm.
Complementarios realizados
Hemoquímica: Hemoglobina: 116 g/L. Hematocrito: 39 %. Leucocitos: 7,3 x 10 8/l. Eritrosedimentación: 35 mm/H. Glicemia: 3,0 mmol/L.
Función hepática: TGP: 7,4 UI. TGO: 4,1 UI.
Función renal: Creatinina: 74,1 µmol/L. Ácido úrico: 341,3 mmol/L.
Lípidos en sangre : Colesterol: 4,48 mmol/L. Triglicéridos: 1,04 mmol/L.
Cituria: Normal.
Heces fecales: Negativa.
Función tiroidea: TSH: 2,1 mu/L. T 4: 135,4 nmol/L.
Ritmo circadiano de cortisol: 11:00 pm: 127 nmol/L. 8:00 am: 355 nmol/L.
Test de estimulación con clonidina
GH: –30 min: 3,1 ng/mL. 0 min: 6,2 ng/mL. 60 min: 3,5 ng/mL. 90 min: 3,9 ng/mL. Valores de referencia: 5-10 ng/mL.
Test de hipoglucemia inducida por insulina
Glicemia: –30 min: 4,73 mmol/L. 0 min: 4,07 mmol/L. 20 min: 3,62 mmol/L. 30 min: 3,23 mmol/L. 60 min: 3,12 mmol/L.
GH: –30 min: 4,5 ng/mL. 0 min: 4,9 ng/mL. 20 min: 5,1 ng/mL. 30 min: 5,6 ng/mL. 60 min: 3,8 ng/mL.
Datos referentes a la madre
Talla: 132,2 cm.
Función tiroidea: TSH: 2,8 mU/L. T3: 1,9 nmol/L. T4: 125,3 nmol/L.
Ritmo circadiano de cortisol: 11:00 pm: 157 nmol/L. 8:00 am: 380 nmol/L.
Test de sensibilidad a la insulina
GH: –30 min: 3,5 ng/mL. 0 min: 2,9 ng/mL. 20 min: 4,0 ng/mL. 30 min: 4,7 ng/mL. 60 min: 4,8 ng/mL.
Glicemia : –30 min: 5,02 mmol/L. 0 min: 5,30 mmol/L. 20 min: 2,16 mmol/L. 30 min: 2,43 mmol/L. 60 min: 4,5 mmol/L.
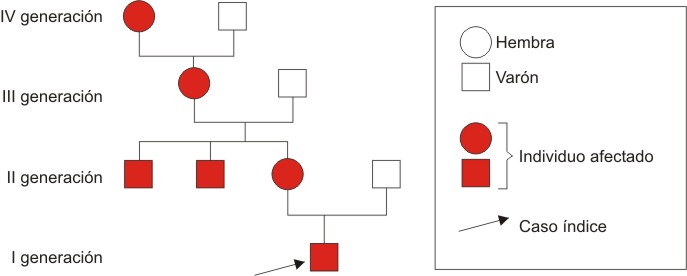
FIG. 3. Árbol genealógico.
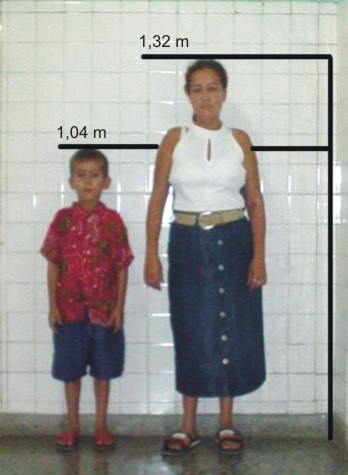
FIG. 4. Madre del paciente con similares características fenotípicas.
Discusión
Dentro de las causas condicionantes de deficiencia de GH, un grupo heterogéneo, en el que se han producido avances significativos recientemente, es el de los defectos hereditarios relacionados especialmente con los progresos en las técnicas recombinantes de ácido desoxirribonucleico (DNA), lo que ha permitido la identificación y organización de la secuencia del gen de la hormona humana de crecimiento y del cluster del gen hGH. Al analizar la variedad etiológica genética que puede alterar la capacidad de producción o acción de la hormona de crecimiento, comprobamos que la ausencia de características clínicas, como una facie peculiar con desproporción craneofacial, frente amplia, apilamiento dental, con falta total de respuesta de GH a las pruebas de estimulación, nos hace descartar la posibilidad de tratarse de un déficit de tipo IA, forma más grave que además se caracteriza por un modo de herencia autosómico recesivo. 10-16 El tipo IB se transmite de igual forma y los niños afectados tienen un retraso del crecimiento menos grave, en el que se obtienen niveles bajos de GH en respuesta a los estímulos.17
Dados los resultados observados en el estudio del paciente, podemos definir el caso como un déficit aislado de hormona de crecimiento, de tipo familiar, y por las referencias de la bibliografía mundial podríamos inferir que se trata de un defecto de tipo II, cuyo patrón de trasmisión es autosómico dominante, lo cual se confirma por el hallazgo de compromiso familiar de forma consecutiva, lo que nos indica el carácter genético y dominante presente en este caso. Es probable que la falla genética sea no sólo en la producción de GH, sino también en los genes que participan en la regulación, secreción y acción de esta, lo cual no es posible comprobar con la tecnología disponible.
Un elemento importante que confirma nuestro diagnóstico presuntivo es la excelente respuesta a la terapia con hormona de crecimiento biosintética (1 unidad subcutánea nocturna, diaria). Al concluir 1 año de tratamiento se obtuvo un incremento marcado de la velocidad de crecimiento, que alcanzó 10,5 cm.
En la mayoría de los pacientes con esta alteración se encuentra una mutación en el intrón III (5955 G-A/C, 5960 T-C y 5982-89 del 5982 G-A) que provoca la pérdida del exón 3 durante el procesamiento del precursor del ARN mensajero que codifica GH1, y origina una proteína truncada. Aunque no se conoce con exactitud el mecanismo por el cual esta proteína inactiva la acción de la GH normal, parece deberse a una alteración del transporte intracelular o a la formación de heterodímeros.18-22 Sin embargo, la tecnología necesaria para hacer estos diagnósticos genéticos aún no está al alcance de la mayoría de los centros de investigación y/o asistenciales, por tratarse de métodos complejos y extremadamente costosos. La literatura cita la reacción en cadena de la polimerasa (PCR), la secuenciación de nucleótidos, la transferencia Southem, entre otros; de ahí que se le conceda tanta importancia a los aspectos auxológicos característicos de este cuadro.
El patrón de respuesta a las pruebas farmacológicas también descarta la posibilidad de tratarse de una GH bioinactiva o también conocido como síndrome de Kowaski, en los que aparecen niveles bajos de IGF-1 pero también respuestas de GH a los estímulos normales o exageradas determinadas por radioinmunoensayo, en contraste con niveles muy disminuidos de GH determinados por radioinmunorreceptor ensayo, a pesar de comportarse clínicamente como un déficit clásico.23,24
Se concluye que el déficit de hormona de crecimiento aislada o asociada a otros déficit hormonales se presenta en 1 de cada 4 000 - 10 000 individuos en la población general. De este grupo se conoce que entre el 5 y el 30 % con retraso grave del crecimiento tiene algún familiar afectado, lo cual sugiere su origen genético. La presencia de un paciente con enfermedad genética constituye una circunstancia que obliga a estudiar la genealogía relacionada con el individuo propósito, con el objetivo de identificar los patrones de transmisión de la enfermedad y dilucidar la presencia de otros sujetos afectos. La incidencia familiar de cualquier entidad, y en especial aquellas que afectan la secreción de GH, exige en la actualidad un estudio clínico y bioquímico exhaustivo de los pacientes para el diagnóstico de la deficiencia hormonal, investigaciones que requieren la aplicación de técnicas de biología molecular para detectar posibles alteraciones en los genes que intervienen en el funcionamiento del eje somatotropo de manera íntegra. En primer lugar, la identificación de estos defectos ha contribuido, de manera notoria en los últimos años, al progreso de la ciencia y la comprensión de los mecanismos fisiopatológicos de múltiples situaciones clínicas. En segundo lugar, permite al facultativo realizar un adecuado enfoque terapéutico, conocer las posibles limitaciones del tratamiento con hormona de crecimiento, orientar la introducción temprana de terapias alternativas, así como el adecuado asesoramiento y consejo genético oportuno destinado a padres y a los mismos pacientes.
Summary
Familial isolated GH deficiency: report of the first case in Cuba
The isolated GH deficiency is an affection of congenital or acquired nature characterised by the partial or total absence of GH detectable in plasma or serum. It is reported the case of an 11-year-old white male patient that received medical attention due to a marked delay of growth and development with height/age corresponding to 4.4 years old and GH figures that were not over 10 ng/mL in pharmacological tests. The rest of the hormonal study was within the normal parameters. Low height was observed in the mother and the hormonal study had similar results. The search for low height individuals in the family showed the same characteristics in the maternal grandmother and great-grandmother. A familial isolated GH deficiency type II with pattern of autosomal dominant inheritance was diagnosed. Treatment with biosynthetic growth hormone was indicated with satisfactory results. It was concluded that the study of genealogy of an index case allows to explain the presence of other affected subjects and to establish the patterns of transmission of the genetic disease .
Key words: Isolated GH deficiency, family deficiency.
Referencias bibliográficas
1. Mericq V, Cassorla F: Sistema hormona del crecimiento-efector y su rol en el crecimiento infantil. Rev Chil Pediatr. 1997;68:27-37.
2. Pombo A, Barreiro J, Fernández Bustillo JM. Etiopatogenia y clasificación del déficit de hormona de crecimiento. Actualizaciones en Endocrinología III. 2002;1-18.
3. Parks JS, Pfaffle RW, Brown MR. Growth hormone deficiency. In: Weintraub BD (ed) . Molecular Endocrinology: Basic. Concepts and Clinical Correlations. New York, Raven Press. 1995.p.473-90.
4. ferrández longás a. Hipocrecimiento por déficit de GH o de sus mediadores. En: 5º Curso de formación de postgrado. Sociedad Española de Endocrinología Pediátrica (Eds). Hipocrecimiento. Palma de Mallorca, 1999:99-112.
5. Castran AO. Características del crecimiento y desarrollo físico. Manual de Pediatría. Madrid McGraw-Hill interamericana de España. 2000.p.1-16.
6. Mahoney CP: Evaluating the child with short stature. Pediatr Clin North Am. 1987;34:825-48.
7. Rosenfeld R, Albertsson-Wikland K, Cassorla F: Diagnostic controversy, the diagnosis of childhood growth hormone deficiency revisited. J Clin Endocrinol Metab.1995;80:1532-40.
8. Carroll PV, Christ ER, Bengtsson BA. Growth hormone deficiency in adulthood and the effects of growth hormone replacement: a review. J Clin Endocrinol Metab. 1998;83:382-395.
9. Bengtsson BA, Johannsson G, Shalet SM, Simpson H, Sonken PH. Therapeutic controversy. Treatment of growth hormone deficiency in adults. J Clin Endocrinol Metab. 2000;85:933-7.
10. Domené HM, Martínez AS, Heinrich JJ. Deficiencia hereditaria de hormona de crecimiento. En: Pombo AM. Tratado de Endocrinología Pediátrica. Madrid: McGraw-Hill interamericana de España. 2002.p.432-42.
11. Codner E, Mericq V, Ugarte F: Utilidad de la determinación del factor de crecimiento insulino símil tipo 1 y de su proteína ligante tipo 3 en el diagnóstico de la deficiencia de hormona de crecimiento en niños. Rev Med Chile. 1999;127:807-13.
12. Pérez Jurado LA, Argente J. Molecular basis of familial growth hormone deficiency. Horm Res. 1994;42:189-97.
13. Parkin JM. Incidence of growth hormone deficiency. Arch Dis Child 1974; 49:904–5.
14. Vimpani GV,Vimpani AF, Lidgard GP. Prevalence of severe growth hormone deficiency. BMJ. 1977;2:427-30.
15. Lindsay R, Feldkamp M, Harris D. Utah Growth Study: growth standards and the prevalence of growth hormone defi ciency. J Pediatr. 1994;125:29-35.
16. López-Bermejo A, Buckway CK, Rosenfeld RG. Genetic defects of the growth hormone-insulin-like growth factor axis. Trends Endocr Metab. 2000;11:39-50.
17. Saggese G, Ranke MB, Saenger P. Diagnosis and treatment of growth hormone deficiency in children and adolescents: toward a consensus. Ten years after the Availability of recombinant human growth hormone workshop held in Pisa, Italy. Hormone Res. 1998;50:320-40.
18. Cogan JD, Phillips III JA, Schenkman SS. Familial growth hormone deficiency: a model of dominant and recessive mutation affecting a monomering protein. J Clin Endocrinol Metab. 1994;79:1261-65.
19. Cogan JD, Ramel B, Lehto M. A recurring dominant negative mutation causes autosomal dominant growth hormone deficiency: a clinical research center study. J Clin Endocr Metab. 1995;80:3591-5.
20. Binder G, Ranke MB. Screening for growth hormone gene splice- sites mutations in sporadic cases with severe isolated GH deficiency using ectopic transcrip analysis. J Clin Endocrinol Metab. 1995;80:1247-52.
21. Cogan JD, Prince MA, Lekhakula S. A novel mechanism of aberrant pre-RNA splicings in humans. Hum Molec Genet. 1997;6:909-12.
22. Wagner JK, Eblé A, Hindmarsh PC, Mullis PE. Prevalence of human GH-1 gene alterations in patients with isolated growth hormone deficiency. Pediatr Res. 1998;43:105-10.
23. Rodríguez HF. Síndrome de crecimiento bioinactiva. Actualizaciones en endocrinología III. 2002.p.55-66.
25. Guyda H: Four decades of growth hormone therapy for short children: what have we achieved? J Clin Endocrinol Metab.1999;84:4307-16.
Recibido: 22 de mayo de 2005. Aprobado: 5 de octubre de 2005.
Dra. Tania Espinosa Reyes . Instituto Nacional de Endocrinología. Zapata y D, Vedado, Ciudad de La Habana, Cuba.
1Especialista de I Grado en Endocrinología.
2Residente de tercer año de Endocrinología.
3Especialista de I Grado en Pediatría.
4Especialista de II Grado en Endocrinología. Hospital Pediátrico del Cerro.
5Doctor en Ciencias Médicas. Especialista de II Grado en Endocrinología. Investigador Titular. Profesor Titular.

