










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Endocrinología
versión On-line ISSN 1561-2953
Rev Cubana Endocrinol vol.23 no.2 Ciudad de la Habana mayo-ago. 2012
REVISIÓN BIBLIOGRÁFICA
Aspectos más recientes en relación con la diabetes mellitus tipo MODY
Most recent aspects about type MODY diabetes mellitus
Dra. Ana Ibis Conesa González, Dra. Teresa Margarita González Calero
Instituto Nacional de Endocrinología. La Habana, Cuba.
RESUMEN
Antecedentes: la mejor comprensión fisiopatológica de la diabetes mellitus permite identificar diferentes tipos, entre ellos una variante monogénica denominada por sus siglas en inglés MODY (Maturity-Onset Diabetes of the Young).
Objetivos: describir los aspectos fisiopatológicos, clínicos, diagnósticos y terapéuticos de los diferentes subtipos MODY.
Desarrollo: los pacientes con diabetes tipo MODY presentan un comportamiento similar a la diabetes mellitus del adulto. Se caracteriza por una alteración genética autosómica dominante inherente y primaria a un defecto en la secreción de insulina. Hasta el momento actual se aceptan 9 subtipos de MODY. Los subtipos 1, 3, 4, 5 y 6 afectan a genes que codifican a factores nucleares de trascripción, y el subtipo 2 al gen que codifica a la enzima glucoquinasa. Se caracterizan clínicamente por cuadros que oscilan entre hiperglucemias permanentes, leves o moderadas, con buen pronóstico clínico, y pocas complicaciones, hasta cuadros de hiperglucemias mantenidas acompañadas de complicaciones crónicas precoces y graves.
Conclusiones: las personas que padecen diabetes tipo MODY no son tan infrecuentes como se piensa. La correcta y temprana identificación de la enfermedad permitirá una acción terapéutica más racional y adecuada para brindar la posibilidad de mejor calidad de vida de estas personas.
Palabras clave: diabetes, MODY, genética, diabetes monogénica, terapéutica.
ABSTRACT
Background: a better physiopathological understanding of diabetes mellitus allows identifying its different types such as a monogenic variant called MODY (maturity-onset diabetes of the young).
Objectives: to describe the physiopathological, clinical, diagnostic and therapeutic aspects of the different subtypes in MODY diabetes.
Development: the patients with MODY diabetes behave similarly to those suffering diabetes mellitus in adults. It is characterized by inherited dominant autosomal genetic alteration which is primary to a defect in insulin secretion. Up to the present, 9 subtypes are accepted. The subtypes 1,3,4,5 and 6 affect gens coding for nuclear transcriptional factors whereas subtype 2 affects the gen coding for glycokinase enzyme. From the clinical viewpoint, they are characterized by conditions ranging from permanent, slight or moderate hyperglycemias, with good clinical prognosis and low complications, to sustained hyperglycemias accompanied by early chronic and serious complications.
Conclusions: the people suffering MODY diabetes are not as uncommon as one might think. The correct and early detection will allow taking quick, adequate and more rational therapeutic actions to provide the patients with a better quality of life.
Key words: diabetes, MODY, genetics, monogenic diabetes, therapeutics.
INTRODUCCIÓN
Atendiendo a los criterios de la Asociación Americana de Diabetes (ADA)1 para la clasificación de la diabetes mellitus (DM), se describe una variante monogénica, denominada Maturity-Onset Diabetes of the Young (por sus siglas en inglés MODY), que se distingue, entre otros aspectos, de las formas más comunes de DM 1 y 2, por la etiología poligénica y multifactorial de estas. MODY se refiere a formas hereditarias de diabetes, causadas por mutaciones en genes autosómicos dominantes, que resultan en un daño en la secreción de insulina. No se refiere a la presencia de una DM 2 en una persona joven, como erróneamente se infiere por su nombre. El término data desde 1964, cuando se consideraba que existían solo 2 tipos de diabetes (insulino y no insulino dependientes), ahora tipos 1 y 2. Fue descrita por vez primera en 1974 por Tattersall (UK)2 y Fajan (USA).3 Afecta aproximadamente del 2 al 5 % de las personas con diabetes, y es frecuentemente tratada como una diabetes tipo 1 o 2.4
Se presenta en individuos jóvenes, usualmente menores de 25 años, y se caracteriza por un defecto en la secreción de insulina que puede ser moderado o severo, dependiendo del gen mutado. El control metabólico de estas personas dependerá de la magnitud del defecto secretor, por lo que puede requerir la administración de insulina de manera similar a los diabéticos tipo 1.5 Es el resultado de mutaciones en distintos genes, en diferentes grupos étnicos. Una sola mutación en un único gen es suficiente para que la enfermedad se exprese, es decir, puede observarse herencia de una sola rama paterna o materna con aproximadamente el 50 % de individuos afectados en 3 generaciones o más. Su patrón de herencia es autosómica dominante. En este tipo de DM la afectación a familiares es superior a la encontrada en otros tipos, se describe que el 85 % de los padres están afectados y el 50 % de los hermanos.6
El modo de herencia y la alta frecuencia de familias con individuos afectados en 3 generaciones facilitaron estudios genéticos (ligamento con marcadores genéticos y clonación posicional) para identificar los genes mutados. Hasta la actualidad se han descrito varios genes implicados, y se espera la identificación de otros. Los defectos en los genes de las proteínas hasta ahora descritos, resultan en la alteración de los procesos de síntesis y secreción de insulina de las células b pancreáticas.7-13
Con esta revisión nos proponemos brindar una información clínico epidemiológica, genética, diagnóstica y terapéutica actualizada sobre la diabetes tipo MODY, lo que permitirá realizar un diagnóstico correcto de una enfermedad subregistrada por la similitud de su cuadro clínico con otros tipos de DM, en aras de contribuir al conocimiento médico y favorecer el tratamiento y pronóstico de estos pacientes.
Características más comunes de la diabetes tipo MODY:14
1. Existe un defecto primario en la función de las células b pancreáticas y déficit en la secreción de insulina.
2. No suele iniciarse con cetosis o cetoacidosis.
3. Tiene una herencia autosómica dominante. Es frecuente encontrar 3 generaciones de una misma familia afectada.
4. Inicia, por lo general, antes de los 25 años, con frecuencia en la infancia o adolescencia.
5. No requiere del uso de insulina al menos en los 5 años posteriores al diagnóstico.
6. No suele asociarse con obesidad o sobrepeso.
7. Suele tener una evolución lenta y progresiva.
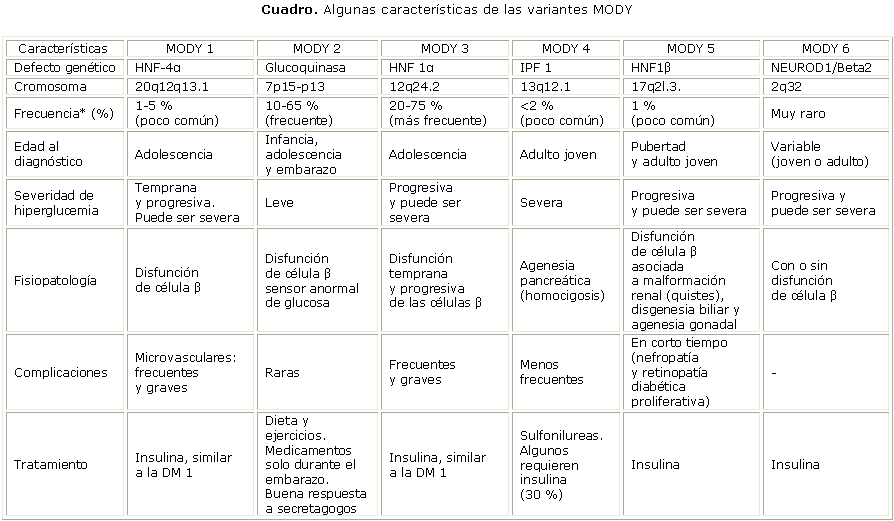
Subtipos de MODY (cuadro)
MODY 1
Es causada por una mutación en el gen HNF-4a, que codifica el factor nuclear de hepatocito 4a (HNF-4a) localizado en el cromosoma 20q12q13.1. El HNF-4a es un miembro de la superfamilia de receptores nucleares, encargados de regular la expresión de distintos genes en: hígado, riñones, intestino e islotes pancreáticos; en estos últimos participa en regular la secreción de insulina en respuesta a la glucosa.15,16 Se han estudiado las propiedades de distintas formas mutantes de este gen MODY, entre ellas, la pérdida en la actividad de transactivación, alteraciones en la dimerización de la proteína, así como alteraciones en la unión de esta proteína al DNA.17
Mutaciones en HNF-4a se asocian a alteraciones en la expresión del transportador de glucosa GLUT 2, de la enzima glucolítica aldolasa B y la gliceraldehído-3fosfato deshidrogenasa, y en el hígado de la enzima piruvato cinasa, esta última disminuye la fase aeróbica de la glucólisis, por lo que reduce así la glucosa y la fuente de energía. Hasta hoy, se han encontrado 8 mutaciones distintas para HNF-4a en pacientes MODY 1: R154X, R127W, V255M, Q268X, G115S, E276Q, K99fsdelAA y F75fsdelT.18-21
Es menos prevalente que los pacientes con MODY 2 y 3, y más frecuente en la población caucásica. Se caracteriza clínicamente por una deficiencia temprana en la insulinosecreción con la consiguiente hiperglucemia, que empeora con el paso del tiempo. Se asocia de forma habitual con complicaciones microvasculares, y rara vez a la obesidad. El diagnóstico se realiza mayoritariamente en adolescentes. Requiere tratamiento con insulina en el 30-50 % de los casos, de manera similar al diabético tipo 1. Este tipo de DM representa el 0,25 % de los diagnosticados inicialmente como DM 2 y el 1-5 % de todos los pacientes con MODY.22,23
MODY 2
Prevalece en las 2 primeras décadas de la vida, sigue en orden de frecuencia al MODY 3. La existencia de esta forma de MODY fue presentada por Froguel y otros,24 quienes realizaron un estudio de ligamiento genético en 16 familias francesas con MODY. La mutación causante de esta entidad se encuentra en el gen que codifica la enzima glucoquinasa, localizado en el cromosoma 7p15-p13. El gen de la glucocinasa humana comprende una región de más de 20 (kilobases) kb y consta de 12 exones.25,26 Esta enzima es la encargada de la fosforilación de la glucosa a glucosa 6-fosfato, primer paso para la glucólisis. Es considerada como el sensor glucémico de la célula b pancreática. Los pacientes tienen incrementado el umbral mínimo para la liberación de insulina, con una secreción retardada en respuesta a glucosa y un menor incremento de glucógeno hepático. Hasta el momento se conocen más de 200 mutaciones que afectan al gen de la glucoquinasa, sin relación con el fenotipo de los afectados. Se han descrito mutaciones inactivantes en homocigosis que causan diabetes neonatal permanente, así como mutaciones activadoras en heterocigosis que causan hipoglucemia hiperinsulinémica persistente en la infancia.27
Es la forma más común en Francia, y representa el 63 % de los casos. Parece ser la forma menos severa de la enfermedad, porque causa solo hiperglucemia leve o reducida tolerancia a la glucosa con buen pronóstico clínico. El diagnóstico se hace con mayor frecuencia en la infancia y la adolescencia. A menudo se detecta por primera vez durante el embarazo, por lo que se confunde el diagnóstico con una diabetes gestacional.28 Pocos casos presentan síntomas, y no necesita tratamiento medicamentoso, excepto en el embarazo. Los pacientes se controlan fácilmente con dieta, lo que hace pensar que este subtipo de DM podría ser más frecuente de lo que se diagnostica.14
Aparece en cada generación, lo que hace pensar que los individuos afectados son hijos de padres afectados (excepto en mutaciones de novo), y el descendiente tiene el 50 % de probabilidades de nacer con la enfermedad. Cuando los 2 progenitores son portadores de mutaciones en el gen GK, la probabilidad se eleva a 75 %. Los grupos étnicos más afectados son caucásicos (con una distribución de 12,5 % entre familias británicas y 63 % en familias francesas), asiáticos (raro en japoneses) e isleños del Océano Pacífico, y es poco frecuente el hallazgo de obesidad y de complicaciones, no tiene predominio por sexo. Presentan una respuesta adecuada a los secretagogos, lo que permite intentar el tratamiento farmacológico con hipoglucemiantes orales.29
MODY 3
Constituye la variedad más frecuente. Se asocia a mutaciones en el gen HNF-1a (factor nuclear del hepatocito-1a), codificado por el cromosoma 12q24.2, y se caracteriza por un defecto primario de la secreción de insulina. Interviene en el desarrollo embrionario y está regulado por sí mismo o por HNF-4a (causante del MODY 1).30 El HNF-1a es esencial para la transcripción del gen de insulina. Formas mutantes de la enzima, con un efecto dominante negativo sobre la actividad de la copia normal, se asocian a una disminución de la expresión del RNAm de la insulina. La expresión de genes involucrados en el transporte y metabolismo de la glucosa incluyen el transportador de glucosa GLUT 2 y la piruvato cinasa tipo L, regulados también por el HNF-1a. La pérdida de la función del HNF-1a lleva a defectos severos en las respuestas secretoras de insulina a la glucosa y leucina.
El HNF-1a es una proteína que se expresa en el hígado, intestino, estómago, riñones y páncreas. Se han identificado más de 80 mutaciones donde la P291fsinsC-HNF resulta ser la más común. La forma funcional es un dímero, un homodímero o un heterodímero con la proteína HNF-1a estructuralmente relacionada, que activa la transcripción de distintos genes. Está compuesto de 3 dominios funcionales: uno amino, el dominio de dimerización terminal (aminoácidos 1-32); un dominio de unión-DNA (aminoácidos 150-280); y un dominio de transactivación carboxilo-terminal (aminoácidos 281-631).29,30
Es el tipo más frecuente en la población adulta de procedencia hospitalaria. Se suele diagnosticar entre los 14 y 30 años, con mayor incidencia durante la adolescencia. La hiperglucemia y el déficit insulínico son severos. Predomina en caucásicos y japoneses. La obesidad no es común. El porcentaje de distribución entre las familias británicas es de 65 %, mientras que en las francesas es de 21 %. Los afectados presentan glucemias normales hasta los 10 años, con deterioro posterior de la función b pancreática, de tal forma que a los 30 años tienen una evidente disminución de la secreción de insulina, que determina la necesidad de tratamiento: dieta, compuestos orales o insulina (pueden requerir tratamiento con insulina de forma muy similar a la DM 1). Las complicaciones microvasculares y neuropáticas son frecuentes y severas.17
Los pacientes con MODY 3 son más sensibles a las sulfonilureas que los diabéticos tipo 2, hecho observado en familias con mutaciones diferentes. Es un ejemplo de farmacogenética, con etiología subyacente a un defecto genético, que altera la respuesta farmacológica al tratamiento.31 En individuos con diagnóstico de MODY 3 se han encontrado valores muy bajos, alrededor de 0,20 mg/L (rango 0,03 a 1,14 mg/L), de proteína C reactiva cardioespecífica (hs-CRP). En la actualidad esta técnica pudiera representar una forma efectiva de screening para este tipo de MODY, pues se encuentra la alteración en el 80 % de los casos.32
MODY 4
Causada por mutaciones en el factor promotor del gen de la insulina 1 (IPF-1), localizado en el cromosoma 13q12.1, implicado en la regulación de la transcripción de insulina, así como en la organogénesis pancreática. En los humanos se ha identificado que mutaciones en IPF-1 producen MODY cuando el gen afectado se encuentra en estado heterocigoto (una sola copia del gen), y agenesia pancreática cuando el gen afectado se encuentra en estado homocigoto (ambas copias del gen mutadas). Tienen menor riesgo de complicaciones crónicas, al compararlos con los subtipos MODY 1, 3 y 5. El diagnóstico se realiza generalmente en adultos jóvenes, y representa por lo menos el 1 % de todos los casos MODY. Tiene prevalencia y penetrancia baja, y solo requiere tratamiento con insulina en el 30 % de los casos.14,33-36
MODY 5
Relacionada con mutaciones en el factor nuclear del hepatocito 1 b (HNF-1b), que se localiza en el cromosoma 17q21.3, es un factor de transcripción nuclear que se expresa en los islotes pancreáticos, y se sugiere que funciona como homodímero o como heterodímero junto a HNF-1a para regular la expresión de distintos genes en diferentes tejidos, particularmente en el páncreas y riñón, afectando la función y organogénesis pancreática y renal. Mutaciones en HNF-1b se asocian a un mayor riesgo para desarrollar enfermedad renal severa.37,38
Al igual que otras variedades MODY, el diagnóstico se realiza antes de los 25 años, y se caracteriza por un defecto secretor de insulina. Representa alrededor del 1 % de los casos MODY. Las características clínicas son variables y se pueden asociar a quistes renales (mutaciones de este gen son una causa importante de enfermedad renal), disgenesia biliar y agenesia gonadal. Los pacientes con mutación en HNF-1a o 1b parecen tener un umbral más bajo para la eliminación de glucosa (que normalmente se considera de 180 mg/dL), lo cual indica que pueden presentar glucosuria más tempranamente que el resto de los diabéticos, y como consecuencia, el desarrollo de complicaciones en menor tiempo.14
La mutación R177X en HNF-1b se asocia frecuentemente con nefropatía y con retinopatía diabética proliferativa, y eventualmente requiere insulina para el control metabólico. Se han descrito otras alteraciones agregadas, como aplasia vaginal y útero rudimentario en 2 mujeres de nacionalidad japonesa. Algunos estudios sugieren que HNF-1b es importante para el desarrollo normal de la unidad anatomo-funcional renal (nefrona) en el feto humano, y cuando se presenta una mutación, tiene como consecuencia el desarrollo de enfermedad renal progresiva.39,40
MODY 6
Se relaciona con mutaciones en el NEUROD1/b2, un factor de transcripción con dos exones, cuyo locus ha sido mapeado en el cromosoma 2q32. El NEUROD1 es una proteína que funciona como regulador del desarrollo pancreático, con expresión tisular en el páncreas, intestino y sistema nervioso central. Malecki y otros (1999) describieron dos mutaciones en NEUROD1, las cuales están asociadas con el desarrollo de la diabetes en el estado heterocigótico. La primera es una mutación "missense" (R111L) del dominio de unión al ADN; y la segunda, es la inserción de una C en un tracto de poli C en el codón 206 del exón 2 (designada como 206+C), lo que produce un cambio en el marco de lectura y un polipéptido truncado que carece del dominio de transactivación carboxilo terminal, una región que se asocia con los coactivadores CBP y p300.14,23
Las mutaciones en NEUROD1 producen DM moderada o severa, en edades de aparición variables. De las 2 familias con mutaciones en NEUROD1 descritas, una (portadora de la mutación 206+6), tiene un fenotipo que se asemeja al producido por alteraciones en HNF-1a (edad de aparición temprana, ausencia de obesidad, secreción de insulina conservada). Todos los afectados tenían niveles bajos de insulina sérica, en 2 de ellos hubo necesidad de tratamiento con insulina, y sus niveles de péptido C fueron indetectables, lo que indica la ausencia de producción de insulina endógena. La segunda familia, portadora de la mutación R111L presentó edades de diagnóstico entre los 30 y los 59 años. Todos los portadores de la mutación fueron obesos, y tenían niveles relativamente altos de insulina sérica en ayuna y 2 horas después de una sobrecarga oral de glucosa niveles más elevados.14,41
Otras formas de MODY41,42
Existen otras variedades de MODY de reciente descubrimiento en las cuales sus características aún no están bien dilucidadas, entre ellos tenemos MODY del 7 al 9. Para MODY 7 el gen afectado es el Kruppel-like factor 11, para el 8 el Bile salt dependent lipase y para el 9 el PAX4.
Diagnóstico
El diagnóstico se debe sospechar ante individuos de cualquier edad con hiperglucemia no cetósica, aunque predomina en la segunda o tercera década de la vida. En los niños se puede confundir con el inicio de una DM 1, aunque difiere de esta por la ausencia de anticuerpos (antiinsulina, antiislotes, anti-GAD) y una herencia dominante. Se diferencia de la DM 2 por la ausencia de resistencia a la insulina. A diferencia de otros tipos de diabetes, cursa con hiperglucemia leve. En general no se asocia a obesidad, pero cuando se presenta se relaciona con menor edad al diagnóstico. La presencia de enfermedad renal puede ser indicativa de MODY 5. Excepto el MODY 2, evolucionan de forma progresiva hacia el fallo de las células beta.42
Características clínicas que sugieren el diagnóstico de MODY:43,44
- Hiperglucemia de ligera a moderada (130-250 mg/dL o 7-14 mmol/L), descubierta antes de los 30 años de edad, aunque cualquier individuo menor de 50 años puede tener este diagnóstico.
- Familiar de primer grado con diabetes de similares características.
- Ausencia de anticuerpos positivos u otro elemento de autoinmunidad personal o familiar (por ejemplo, tiroiditis).
- Persistencia de bajos requerimientos de insulina, inferior a 0,5 ud/kg/día), pasado el período de "luna de miel".
- Ausencia de obesidad (no se excluye el diagnóstico en el sobrepeso y obeso) u otros elementos asociados a la DM 2 o al síndrome metabólico (hipertensión arterial, hiperlipidemia, síndrome de ovarios poliquísticos).
- La resistencia a la insulina aparece en raras ocasiones.
- Presencia de enfermedad renal personal o familiar de primer o segundo grado.
- Diabetes neonatal no transitoria, o una aparente DM 1 con inicio antes de los 6 meses de edad.
- Presencia de adenoma hepático o carcinoma hepatocelular, sugiere MODY 3.
- Presencia de enfermedad renal, útero rudimentario o bicorne, aplasia vaginal, ausencia de vasos deferentes sugieren MODY 5.
El test genético establece el diagnóstico basado en un análisis de sangre. Es además útil para el screening de familiares, que puede detectar mutaciones asociadas al MODY en individuos todavía no diabéticos, en los que el incremento de la vigilancia y cambios en el estilo de vida pueden ayudar a prevenir la persistencia de una hiperglucemia no reconocida, además de facilitar la selección de un tratamiento específico con mejor pronóstico. Está indicado su uso en individuos de cualquier edad con hiperglucemia no cetósica, así como en personas con historia familiar de diabetes tipo MODY.45,46
La metodología que se sigue se basa en aislar el ADN de leucocitos y amplificar por reacción en cadena de la polimerasa (PCR) los exones a estudiar (de los que se conocen mutaciones). Estos fragmentos amplificados se analizan, teniendo el inconveniente de un 5 % de falsos negativos. Los fragmentos mutados se secuencian a posteriori para conocer exactamente la mutación que provoca el fenotipo observado. A menudo es difícil el diagnóstico del gen implicado, ya que los factores de transcripción afectados pueden estar interrelacionados. Por ejemplo, HNF-3b regula a HNF-4a (a su vez este regula HNF-1a y HNF-1b), IPF-1 y NEUROD1, que tienen distintas dianas que afectan el metabolismo, el desarrollo de células beta o el recambio proteico, y por eso pueden ser causantes de MODY.47,48
Tratamiento49-53
La perspectiva clínica y el tratamiento para la diabetes tipo MODY depende de la causa genética. El MODY 2 puede ser manejado con alimentación y ejercicios físicos con excelente pronóstico. Los subtipos 1, 3 y 4 responden bien al uso de sulfonilureas, aunque una importante proporción de ellos (30-40 % para MODY 1 y MODY 3), requieren eventualmente terapia insulínica, debido al fallo progresivo de las células beta. El MODY 5 requiere reemplazo de la función pancreática endocrina y exocrina, y tratamiento para múltiples anomalías orgánicas. En los pacientes con sobrepeso la pérdida de peso mejora la hiperglucemia.
Se concluye señalando que la diabetes tipo MODY tiene una herencia autosómica dominante, con un riesgo incrementado de padecerla familiares de afectados. Representa un por ciento no despreciable dentro de la DM, frecuentemente no diagnosticada. Existen una serie de características bien identificadas, descritas en la presente revisión, que nos hacen pensar cómo llegar al diagnóstico de esta entidad. Hasta la actualidad se han descrito 9 tipos con características específicas, lo cual hace que el tratamiento y pronóstico varíen según el subtipo. Resulta importante el conocimiento de esta entidad, y pensar en ella al momento del diagnóstico de una persona con diabetes e historia familiar de varias generaciones afectadas, hace que el tratamiento impuesto sea acertado con un mejor pronóstico y calidad de vida.
REFERENCIAS BIBLIOGRÁFICAS
1. Expert Committee on the Diagnosis and Classification of Diabetes Mellitus: Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 2000;23(Suppl 1):4-19.
2. Tattersal RB. Mild familial diabetes with dominant inheritance. Quarterly Journal of Medicine. 1974;43:339-57.
3. Tattersall RB, Fajans SS. A difference between the inheritance of classical juvenile-onset and maturity-onset type diabetes of young people. Diabetes. 1975;24:44-8.
4. Bellanne-Chantelot C, Dubois-Laforgue D, Velho G. Diagnosis and management of maturity-onset diabetes of the young. Timsit Treat Endocrinol. 2005;4(1):9-18.
5. Evans JC, Bulman MP, Pearson E, Allen L, Owen K, Bingham C, et al. Beta Cell Genes and Diabetes; Molecular and Clinical Characterization of Mutations in Transcription Factors. Diabetes. 2001;50(Suppl 1):S94-100.
6. Doria A, Plengvidya N. Recent advances in the genetics of maturity-onset diabetes of the young and other forms of autosomal dominant diabetes. Curr Opin Endo Diab. 2000;7:203-7.
7. Florez JC, Hirschhorn J, Altshuler D. The inherited basis of diabetes mellitus: implications for the genetic analysis of complex traits. Annu Rev Genomics Hum Genet. 2003;4:257-91.
8. Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med. 2001;345:971-80.
9. Giuffrida FM, Reis AF. Genetic and clinical characteristics of maturity-onset diabetes of the young. Diabetes Obes Metab. 2005;7:318-26.
10. Rubio Cabezas O. Diabetes mellitus monogénica. An Pediatr (Barc). 2008;68(Supl 1):73-7.
11. Velho G, Robert JJ. Maturity-onset diabetes of the young (MODY): genetic and clinical characteristics. Horm Res. 2002;57(Suppl 1):29-33.
12. Hattersley A, Bruining J, Shield J, Njolstad P, Donaghue K, International Society for Pediatric and Adolescent Diabetes. ISPAD Clinical Practice Consensus Guidelines 2006-2007. The diagnosis and management of monogenic diabetes in children. Pediatr Diab. 2006;7:352-60. Erratum in: Pediatr Diab. 2007;8:49-52.
13. Nakhla M, Polychronakos C. Monogenic and other unusual causes of diabetes mellitus. Pediatr Clin North Am. 2005;52:1637-50.
14. Sánchez-Reyes L, Fanghanel G, Márquez-Cid ME, Salazar Rocha R, Labastida-Sánchez C, Solís-Pérez A. Actualización en los diferentes subtipos de diabetes tipo MODY. Rev Endocrinol Nutr. 2001;9:5-11.
15. Lindner T, Gragnoli C, Furuta H, Cockburn BN, Petzold C, Rietzsch H, et al. Hepatic function in a family with a nonsense mutation (R154X) in the hepatocyte nuclear factor-4 alpha/ MODY1 gene. J Clin Invest. 1997;100:1400-5.
16. Shih DQ, Dansky HM, Flaisher M, Assmann G, Fajans SS, Stoffel M. Genotype/phenotype relationships in HNF- 4alpha/MODY1: haploinsufficiency is associated with reduced apolipoprotein (AII), apolipoprotein (CIII), lipoprotein (a) and triglyceride levels. Diabetes. 2000;49:832-7.
17. Wang H, Maechler P, Antinozzi PA, Kerstin A, Hagenfeldt KA, Wollheim CB. Hepatocyte Nuclear Factor 4 Regulates the Expression of Pancreatic-Cell Genes Implicated in Glucose Metabolism and Nutrient-induced Insulin Secretion. J Biol Chem. 2000;275:53-9.
18. Bogan AA, Dallas-Yang Q, Ruse MD, Maeda Y, Jiang G, Nepomuceno L, et al. Analysis of protein dimerization and ligand binding of orphan receptor HNF-4alpha. J Mol Biol. 2000;302:831-51.
19. Lorna W, Harries J, Locke B, Neil A, Hanley K, Steele A, et al. The Diabetic Phenotype in HNF4A Mutation Carriers Is Moderated By the Expression of HNF4alpha Isoforms From the P1 Promoter During Fetal Development. Diabetes. 2008;57:1745-52.
20. Lausen J, Thomas H, Lemm I, Bulman M, Borgschulze M, Lingott A, et al. Naturally occurring mutations in the human HNF-4a gene impair the function of the transcription factor to a varying degree. Nucleic Acids Res. 2000;28:430-7.
21. Velho G, Froguel P. Genetic, metabolic and clinical characteristics of maturity onset diabetes of the young. Eur J Endocrinol. 1998;138:233-8.
22. ISPAD. Consensus guidelines for the manegement of type 1 diabetes mellitus in children and adolescents. Zeist, Netherlands: Medical Forum international; 2000.
23. Froguel P, Vaxillaire M. Close linkage of glucokinase locus on chromosome 7p to early-oset non-insulin-dependent diabetes mellitus. Nature. 1992;356:162-4.
24. Doria A, Plengvidya N. Recent advances in the genetics of maturity-onset diabetes of the young and other forms of autosomal dominant diabetes. Curr Opin Endo Diab. 2000;7:203.
25. Stenson PD, Ball E, Howells K, Phillips A, Mort M, Cooper DN. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577-81.
26. Gloyn AL. Glucokinase (GCK) mutations in hyper and hypoglycemia: maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemia of infancy. Hum Mutat. 2003;22:353-62.
27. Timsit J, Bellanne-Chantelot C, Dubois-Laforgue D, Velho G. Diagnosis and management of maturity-onset diabetes of the young Treat Endocrinol. 2005;4(1):9-18.
28. Cuesta AL. Diabetes monogénica. En: Gomis R, Rovira A, Felíu JE, Oyarzábal M (eds.). Tratado SED de Diabetes Mellitus. Bases moleculares clínicas y Tratamiento. Madrid: Ed. Panamericana; 2007. p. 47-56.
29. Pearson ER, Liddel WG, Shephar M, Corral R, Hattersley AT. Sensitivity to sulphonilureas in patients with hepatocyte nuclear factor-1a gene mutation: evidence for pharmacokinetics in diabetes. Diabet Med. 2000;17:543-45.
30. Marcondes Lerario A, Pinto Brito L, Marinho Mariani B, Candida Fragoso M, Cesar Machado MA, Teixeira R. A missense TCF1 mutation in a patient with MODY-3 and liver adenomatosis. Journal List Clinics (Sao Paulo) [serie en Internet]. 2010 [citado 26 de noviembre de 2011];65(10). Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2972616/
31. Tomasz Klupa. Determinants of the Development of Diabetes (Maturity-Onset Diabetes of the Young-3) in Carriers of HNF-1 (alpha) Mutations Evidence for parent-of-origin effect. Diabetes Care. 2002;25:2292-301.
32. Katharine R. Owen. Assessment of High-Sensitivity C-Reactive Protein Levels as Diagnostic Discriminator of Maturity-Onset Diabetes of the Young Due to HNF1A Mutations. Diabetes Care. September 2010;33(9):1919-24.
33. Stoffers DA, Ferre J, Clarke WI, Habaner JF. Early-onset type II diabetes mellitus (MODY) linked to IPF1. Nat Genet. 1997;15:106-10.
34. Giuffrida FM, Reis AF. Genetic and clinical charactericts of maturity-onset diabetes of the young. Diab Obes Metab. 2005;7:318-26.
35. Stoffers DA, Zinkin NT, Stanojevic V. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. 1997;15:106.
36. Stoffers DA, Ferrer J, Clarke WL. Early-onset type II diabetes mellitus (MODY 4) linked to IPF1. Nat Genet. 1997;17:138-40.
37. Horikawa Y, Iwasaki N, Hara M. Mutation in hepatocyte nuclear factor-1 (beta) gene (TCF2) associated with MODY. Nat Genet. 1997:17:384-5.
38. Nishigori H, Yamada S, Kohama T, Tomura H, Sho K, Honikawa Y. Frameshift mutation, A263fsinsGG, in hepatocyte nuclear factor-1 beta gene associated with diabetes and renal dysfunction. Diabetes. 1998;47:1354-55.
39. Iwasaki N, Ogata M, Tomonaga O, Kuroki H, Kasahara T, Yano N, et al. Liver and kidney function in Japanese patients with maturity-onset diabetes of the young. Diabetes Care. 1998;21:2144-8.
40. Malecki MT, Jhala US, Antonellis A, Fields L, Doria A, Orban T. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Get. 1999:323-6.
41. MODY (Report) [homepage en Internet]. Case report. Retrieved Jan 25; 2010 [citado 26 de noviembre de 2011]. Disponible en: http://www.Phlaunt.com/diabetes/14047009.php.
42. Maturity Onset Diabetes, Spark People [homepage en Internet]. Retrieved Jan 21; 2010 [citado 26 de noviembre de 2011]. Disponible en: http://www.Sparkpeople.com/resource/health_a-z_ detail.asp
43. MODY (Report). Harvard [homepage en Internet]. Case report. Retrieved January 23; 2010 [citado 26 de noviembre de 2011]. Disponible en: http://www.ncbi.nlm.nih.gou/pmc/articles/PMC2972616/
44. Renal Cysts and Diabetes Syndrome (Report) [homepage en Internet]. Case report. Retrieved May 19; 2011 [citado 26 de noviembre de 2011]. Disponible en: http://www.omim.org/entry/137920?search=48.%09Renal%20Cysts%20and%20Diabetes%20Syndrome&highlight=cysts%20diabete%20syndrome%2048%20syndromic%20diabetic%20cyst%20renal%2048.%09renal%20diabetes
45. Fajans S, Bell G, Polonsky K. Molecular mechanisms and Clinical Pathophysiology of Maturity Onset Diabetes of the Young. N Engl J Med. 2001;345(13):971-80.
46. Permutt MA, Hattersley A. Searching for Type 2 diabetes Genes in the Post-genoma era. Trends Endocrinol Metab. 2000;11:383-93.
47. Dhavendra Kumar, Weatherall DJ. Genomics and clinical medicine. Oxford: University Press US; 2010. p. 184.
48. Tusié LMT. La genética de la diabetes mellitus tipo 2: genes implicados en la diabetes de aparición temprana. Revista de Investigación Clínica. 2000;52:296-305.
49. Plengvidhya N, Kooptiwut S, Sontawee N, Dol A, Furuta H, Nishi M, et al. PAX4 mutations in Thais with maturity onset diabetes of the young. J Clin Endocrinol Metab. 2007;92:2821-6.
50. Shimomura H, Sanke T, Hanabusa T, Tsumoda K, Furuta H, Nanjo K. Nonsense mutation of islet-1 gene (Q310X) found in a type 2 diabetic patient with a strong family history. Diabetes. 2000;49:1597-600.
51. Raeder H, Johanssons S, Hola PI, Haldorsen IS, Mas E, Sbarre V, et al. Mutations in CEL VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nat Genet. 2006;38:54-62.
52. Neve B, Fernández-Zapico ME, Ashkenazi-Katalan V, Dina C, Hamid YH, Joly E, et al. Role of transcription factor KLF11 and its diabetes-associated gene variants in pancreatic beta cell function. Proc Nat Acad Sci. 2005;102:4807-12.
53. Ewan R. Pearson. Switching from Insulin to Oral Sulfonylureas in Patients with Diabetes Due to Kir 6.2 Mutations. NEJM. 2006;355(5):467-77.
Recibido: 6 de diciembre de 2011.
Aprobado: 18 de marzo de 2012.
Ana Ibis Conesa González. Instituto Nacional de Endocrinología. Calle Zapata y D, Vedado, municipio Plaza de la Revolución. La Habana, Cuba. Correos electrónicos: ana.conesa@infomed.sld.cu teregonzalez@infomed.sld.cu

{kind=link}