










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Cubana de Endocrinología
versão On-line ISSN 1561-2953
Rev Cubana Endocrinol vol.24 no.2 Ciudad de la Habana maio-ago. 2013
ARTÍCULO ORIGINAL
Estudio preliminar sobre el tratamiento con hormona de crecimiento humana recombinante en el síndrome de Turner
Preliminary study on the recombinant human growth hormone treatment of Turner's syndrome
Dra. Tania Mayvel Espinosa Reyes,I Dr. Lázaro Aramís Pérez Sampert,I Dr. Miguel Ángel Martínez Morales,II Dr. C. Francisco Carvajal MartínezI
IInstituto Nacional de Endocrinología (INEN). La Habana, Cuba.
IIMINSAP. La Habana, Cuba.
RESUMEN
Introducción: los pacientes con síndrome de Turner presentan una monosomía parcial o total del gonosoma X, disgenesia gonadal, diversos rasgos físicos típicos, baja talla e infantilismo sexual.
Objetivo: evaluar el efecto del tratamiento con hormona de crecimiento recombinante sobre la talla en las pacientes con el diagnóstico clínico y cromosómico de síndrome de Turner.
Métodos: se realizó un estudio de tipo descriptivo y retrospectivo a pacientes con síndrome de Turner atendidas en consultas externas en el Departamento de Endocrinología Pediátrica del Instituto Nacional de Endocrinología, desde mayo de 2003 hasta mayo de 2004. La muestra estuvo constituida por 19 niñas. Se confeccionaron dos grupos, uno con aquellas pacientes que recibieron tratamiento con hormona de crecimiento recombinante (n= 9) a una dosis de 0,14 UI/kg/día, administrada vía subcutánea entre las 8:30 y 9:30 pm, que se conoció como grupo A. El segundo quedó conformado con las pacientes que no recibieron dicha hormona (n= 10), denominado grupo B. Los datos necesarios para la investigación fueron obtenidos de la revisión de las historias clínicas.
Resultados: un incremento de la talla en el grupo A, que inició el estudio con una talla basal media de 131,7 ± 7,5 cm, para alcanzar una talla media, después de un año de tratamiento, de 137,9 ± 7,1 cm, con una velocidad de crecimiento media en ese año de 6,2 ± 2,3 cm/año. La comparación de ambos grupos después de un año de estudio mostró diferencias significativas en la talla media al año (p= 0,0071) y la velocidad de crecimiento media al año (p= 0,0032).
Conclusiones: el tratamiento con hormona de crecimiento recombinante durante el primer año resultó efectivo, al acelerar significativamente la velocidad de crecimiento en las niñas con síndrome de Turner. La ganancia de peso corporal resultó adecuada durante el periodo de estudio, pues se logró mantener una valoración nutricional estable sin modificaciones en el canal percentilar. La inducción de la pubertad no cambió el pronóstico de la talla al final del estudio.
Palabras clave: baja talla, síndrome de Turner, hormona de crecimiento.
ABSTRACT
Introduction: Turner's syndrome patients present with total or partial monosomy of X gonosome, general disgenesia, several typical physical traits, short height and sexual infantilism.
Objective: to evaluate the effect of recombinant human growth hormone-based treatment on the height of patients clinically and chromosomally diagnosed as Turner's syndrome subjects.
Methods: a retrospective and descriptive study was conducted in Turner's syndrome patients, who had been seen from May 2003 to May 2004 at the pediatric endocrinology department of the National Institute of Endocrinology. The sample was made up of 19 girls divided into 2 groups. Group A comprised patients who were treated with recombinant human growth hormone (n= 9) at a dose of 0.14 IU/kg/day subcutaneously administered from 8:30 to 9:30 pm. Group B included the patients who did not receive this treatment (n= 10). The required data for the research stemmed from the medical history check-ups.
Results: increase of the Group A patients' height, whose mean basal height at the beginning of the study was just 131.7 ± 7.5 cm and after one year of treatment, they reached 137.9 ± 7.1 cm, at a rate of average growth of 6.2 ± 2.3 cm/year. The comparison of both groups after one year showed significant differences in mean height (p= 0.0071) and mean growth a year (p= 0.0032).
Conclusions: the treatment of these patients with the recombinant growth hormone during the first year was effective, since it markedly accelerated the rate of growth in girls with Turner's syndrome. The body weight gain proved to be adequate in the study period, because it managed to keep steady nutritional assessment without changes in the percentile canal. Inducement to puberty did not alter the final height prognosis at the end of the study.
Key words: short height, Turner's syndrome, human growth hormone.
INTRODUCCIÓN
Es conocido que los humanos necesitan de un cromosoma X intacto para sobrevivir; cuando el segundo cromosoma X o Y se ausenta resulta en un fenotipo prenatal y posnatal característico. Otto Ullrich observó, por vez primera en 1930, una niña de 8 años que presentaba cuello ancho, cubitus valgus, edema linfangiectásico congénito, orejas displásicas y grandes, ptosis palpebral, micrognatia, displasia ungueal y mamilas hipoplásicas. En 1938, en la universidad de Oklahoma, el profesor Henry Turner describió 7 casos de infantilismo y cúbitos valgus.1
Prevalencia
En general, se considera que la aparición del síndrome de Turner (ST) es esporádica, sin tendencia a recurrir, aunque se han descrito casos familiares. La prevalencia descrita es de 1:2 000 a 1:5 000 en recién nacidos vivos mujeres. Se conoce que en el 1 % de todos los embarazos el producto de la concepción presenta una monosomía X, de ellos un alto porcentaje termina en abortos espontáneos en el primer trimestre del embarazo, solo aquellos fetos con "formas moderadas" de ST son viables.1
Características clínicas
La clínica varía según la edad y la anomalía citogenética que presenten. Las características más comunes descritas en el ST son las alteraciones somáticas, el infantilismo sexual y la baja talla. Nos dedicaremos especialmente a detallar aspectos relacionados con el crecimiento.
Con alta frecuencia se recoge la historia de un crecimiento intrauterino retardado, el peso medio aproximado al nacer es de 2 800 g. La velocidad de crecimiento en los 3 primeros años de vida es relativamente normal, y la disminución progresiva de esta ocurre en la etapa escolar, haciéndose más evidente en la adolescencia por la ausencia del estirón puberal, y culmina con una talla final por debajo del percentil 3. Durante la adolescencia el crecimiento es lento, se observa un retardo en la maduración ósea y un cierre epifisario tardío que se señala entre 18 y 20 años. Esto último favorece la terapia con hormona de crecimiento (hGH) por un tiempo prolongado.
La talla final en la curva de Lyon2 en pacientes con ST sin tratamiento es de 142,9 ± 7,3 cm, obtenida en investigación conjunta de EUA y Europa por más de 20 años. Se han estudiado algunos factores que intervienen en la adquisición de la talla final en las pacientes con ST, y se han señalado el cariotipo, la talla paterna, la etnia, la pubertad espontánea y el inicio temprano del tratamiento con estrógenos, entre los elementos de mayor importancia.
Padrón RS y otros estudiaron 29 pacientes cubanas con ST, de ellas 18 mostraron una fórmula cromosómica 45X, y se observó que todas las niñas que presentaron dicha fórmula se asociaban a baja talla.3 En cuanto al cariotipo, las pacientes con fórmula cromosómica 45X/46XX alcanzan mayor talla; el efecto sobre la talla de otros mosaicismos no está aún establecido.4 Por otra parte, en la mayoría de las pacientes en las que se verifica una pubertad espontánea, esta se asocia a un ligero estirón puberal.5,6
Se dio un gran paso cuando en 1997 el grupo de Heidelberg descubrió el gen short homeobox containing gene (SHOX). El cromosoma X posee en la parte distal de su brazo corto (Xp) una pequeña zona denominada PAR 1 (pseudo autosomic region). Normalmente, en la pareja XX, uno de los cromosomas X está inactivado: se podría pensar entonces que no sirve para nada, pero si fuera así, ¿por qué existiría el ST? Es preciso concebir que una parte del cromosoma X escapa a esta inactivación. Este razonamiento condujo al descubrimiento del gen SHOX, y a la noción de que la región PAR 1 escapaba a la inactivación fisiológica, explicando la talla baja, consecuencia de una haploinsuficiencia (mutación o deleción) de este gen SHOX. Por último, el gen SHOX puede faltar en algunas familias de talla baja constitucional.7-10
El desarrollo óseo patológico se produce por un defecto intrínseco en la osificación, secundario a la pérdida de genes críticos en el cromosoma X. La mayoría de las pacientes con ST no tienen una deficiencia clásica de hormona de crecimiento (GH) demostrada por pruebas de provocación. En una investigación realizada por el grupo italiano de estudio de ST, que incluyó 301 pacientes, se comprobó que el 36,2 % tuvo una reducción en la reserva de GH en 2 pruebas de estimulación (GH< 10 ng/L) y en el 61,8 % una disminución de la media de secreción espontánea nocturna (GH< 3,3 ng/L). No se encontró relación entre estas respuestas y el cariotipo, ni con la aparición espontánea de telarquia y/o pubarquia.11
Algunas pacientes muestran una clara disminución de la velocidad de crecimiento, por lo que se sugiere pesquisar hipotiroidismo o déficit clásico de GH.12 En adolescentes se han comprobado bajos niveles de GH en 24 horas, así como una disminución de amplitud y frecuencia de los pulsos de secreción.13 El fallo del crecimiento es evidente desde la etapa escolar, incluso antes de que el déficit de GH o IGF-1 sea demostrable, lo cual hace pensar en un trastorno intrínseco del hueso más que en una anomalía endocrina como defecto primario.12 En los últimos años se ha instituido el tratamiento con hGH con el fin de mejorar el pronóstico de la talla final de estas pacientes. Sin embargo, los resultados son controversiales, ya que no se obtiene similar respuesta a la estimulación en todos los pacientes.11,13-18
Principales estudios
El estudio pionero, y que por primera vez comunicó la existencia de una mejoría significativa de la talla final, en pacientes con ST bajo tratamiento hormonal, fue el ensayo multicéntrico norteamericano. En él se analizaron dos modalidades terapéuticas: hGH sola (n= 17) o asociada a oxandrolona (n= 43), alcanzándose resultados significativos en la talla final media en ambos grupos. En el grupo que recibió hGH sola, la talla final media fue de 150,4 ± 5,5, con una ganancia neta media de 8,4±4,0 cm; y en la segunda modalidad terapéutica mostró una talla final media de 152,1±5,9, con una ganancia media de 10,3 ± 4,7 cm.19
La experiencia sueca es útil, ya que compara diferentes regímenes de tratamiento con hormona de crecimiento recombinante (rhGH) en función a la asociación o no de oxandrolona y etinilestradiol, con resultados hasta la talla final. Se reportan mejores resultados en el grupo con rhGH más oxandrolona desde el inicio, a una edad ósea de 10 años, logrando una talla final de 154,2 ± 6,6 cm y una ganancia de talla de 8,5 ± 4,6 cm.11 Haeusler y otros estudiaron a 20 pacientes tratadas inicialmente con hGH, y añadiendo tras 2 años oxandrolona, hasta la talla final. La edad cronológica media de inicio de tratamiento fue de 11,8 años, y la duración media de tratamiento 5,7 años. Con estas condiciones, la talla media final fue de 152,9 ± 3,5 cm, y los buenos resultados quedan reflejados por una ganancia neta de talla de 9,3 cm.19
El estudio multicéntrico centroeuropeo analizó la talla final de 79 pacientes tratadas con rhGH en dosis de 0,8 ± 0,2 UI/kg/semana a una edad inicial mínima de 10 años y durante un período de 5 años. Este grupo terminó con una talla final de 150,2 ± 5,2 cm. A pesar de conseguir una talla final buena, su ganancia fue muy variable, desde nula hasta 12,1 cm, siendo la media de 2,9 ± 3,6 cm para los estándares de Ranke.19
El estudio con resultados más importantes en cuanto a la talla final es el holandés. En él se comparan 3 grupos de pacientes con hGH y esquemas de tratamiento diferentes, con una edad de inicio de 6 a 7 años, y con una desviación estándar de talla semejante al inicio. Grupo I (n= 23), 4 UI/m2/día; grupo II (n= 23), 4 UI/m2/día durante el primer año, 6 UI/m2/día posteriormente; grupo III (n= 22), 4 UI/m2/día durante el primer año, 6 UI/m2/día en el segundo y 8 UI/m2/día posteriormente. Tras un período medio de 7,3 años de tratamiento, han publicado unos resultados preliminares muy buenos, en términos de talla final y de ganancia media de talla. La talla media final y la ganancia media de talla de cada grupo son las siguientes: grupo I (n= 10), 158,8 ± 7,1 cm y 12,5 ± 2,1 cm; grupo II (n= 10), 161,0 ± 6,8 cm y 14,5 ± 4,0 cm; y grupo III (n= 12), 162,3 ± 6,1 cm y 16,0 ± 4,1 cm.19
En febrero de 1990 la Comisión de Sanidad de la Comunidad Económica Europea en Bruselas decidió incluir dentro de los criterios de utilización de la hormona de crecimiento a este síndrome.20 Los datos históricos de crecimiento en el ST, así como la talla final, han sido suficientes para poder comparar los resultados del tratamiento con hGH en este síndrome. Por los estudios publicados, estas niñas incrementan aproximadamente en 8 cm la media de su talla final. En esta enfermedad se podrían utilizar otros tratamientos combinados promotores del crecimiento, tales como la oxandrolona y el etinil-estradiol, pero utilizando solo la GH se obtienen buenos resultados terapéuticos.20
Si bien son conocidos los beneficios de la terapia con GH en la adquisición de la talla final de las pacientes con ST, los cuales no se logran con el tratamiento estrogénico sustitutivo exclusivamente, nos propusimos en este estudio evaluar el efecto de la rhGH sobre la velocidad de crecimiento, el peso corporal y la talla después del primer año de tratamiento en un grupo de pacientes con esta entidad. Se presentan los primeros resultados cubanos que ratifican las ventajas de la terapia con hormona de crecimiento en pacientes con ST.
MÉTODOS
Se realizó un estudio descriptivo y retrospectivo de las pacientes con ST atendidas en consulta externa en el Departamento de Endocrinología Pediátrica del Instituto Nacional de Endocrinología, desde mayo de 2003 hasta mayo de 2004. Se llevó a cabo una revisión de las historias clínicas de las pacientes con ST y se conformaron 2 grupos, uno con aquellas que llevaron tratamiento con rhGH (Grupo A, n= 9) a razón de 0,14 UI/kg/día administrada entre las 8:30 y 9:30 PM, subcutánea; y otro, con pacientes que no habían tenido tratamiento con rhGH (Grupo B, n= 10). Por tratarse de un estudio retrospectivo no fue posible asegurar la asignación aleatoria de los grupos que conformaron la muestra, por lo que pretendimos con el mismo un acercamiento al problema.
Se tomó como criterio de inclusión toda paciente con diagnóstico clínico y cromosómico de ST, que fuera atendida en el periodo establecido para la investigación. Un grupo recibió tratamiento con rhGH por un año y el otro no. Como criterio de exclusión se tuvo en cuenta a las pacientes con otras enfermedades crónicas capaces de afectar el crecimiento, con la excepción del hipotiroidismo compensado.
Se realizó inicialmente una descripción de las características de los sujetos, para comprobar si los 2 grupos formados eran comparables, para lo cual se evaluó si eran similares respecto a las variables siguientes:
- Peso (g) y talla (cm) al nacer
- Edad cronológica al inicio del estudio (en años y meses cumplidos)
- Presencia de disfunción tiroidea
- Presencia de déficit de GH (prueba de sensibilidad a la insulina y prueba de clonidina)21-23
- Diagnóstico cromosómico (obtenido por análisis del cariotipo)
Las pacientes fueron talladas y pesadas por una enfermera del departamento, entrenada con ese propósito, utilizando siempre el mismo tallímetro y pesa (las pacientes fueron pesadas sin zapatos y solo con ropa interior). El seguimiento se garantizó por los 4 médicos que integran el Servicio de Endocrinología Pediátrica. Se comparó la talla basal y al año en ambos grupos y entre estos, así como el ritmo de crecimiento en el año previo al estudio, y después de transcurrido un año en ambos grupos y entre ellos, calculándose mediante la fórmula: talla al final del año menos talla al inicio del año. Se describieron los centímetros crecidos en un año previo al inicio del estudio en los grupos A y B, se compararon con los cm crecidos durante el período de la investigación, y se consideró el incremento en más de 2 cm respecto a la velocidad de crecimiento basal como un resultado positivo.
Se determinó la talla diana (Td)24-27 según la talla de los padres. Se recogieron las variables: talla del padre (tp) y talla de la madre (tm), y se empleó la fórmula siguiente:
Se describió la media del peso basal y al año del estudio en ambos grupos y se compararon entre sí, fueron llevados a la curva de percentiles según la relación peso para la edad, y se determinó la relación peso para la talla. Se usaron la curva y la tabla cubanas. Se dieron a conocer las pacientes que durante el estudio recibieron tratamiento con estrógeno y la dosis utilizada. Además, se calculó la edad cronológica media al inicio de este tratamiento.
Los datos necesarios para la investigación fueron obtenidos de la revisión de las historias clínicas, fueron recogidos en una planilla de vaciamiento y posteriormente introducidos en una base de datos elaborada a tal efecto, empleando el programa Microsoft Excel 2000 para facilitar su procesamiento estadístico. Los resultados fueron expresados utilizando como indicadores valores absolutos, media, mediana y porcentajes. La comparación de las variables cuantitativas para ambos grupos se realizó mediante la prueba no paramétrica de Wilcoxon Mann-Whitney para la comparación de valores medianos de muestras independientes. En el caso de la comparación del mismo grupo en diferentes momentos, se empleó la prueba de los signos con rango de Wilcoxon. En las variables cualitativas se evaluó la presencia de asociación estadística mediante el test chi cuadrado. En todos los casos se fijó un nivel de significación de 0,05 y el apoyo en el paquete estadístico SPSS para Windows versión 10,0. Para la confección del informe final se utilizó el editor de texto Microsoft Word 2000 y se presentaron los resultados de la investigación mediante textos, tablas, gráficos y figuras, los cuales permitieron emitir las conclusiones. En las historias clínicas de los pacientes que recibieron tratamiento con rhGH se argumenta acerca de la charla que tuvo el médico y el tutor del paciente sobre los riesgos y beneficios de este tratamiento.
RESULTADOS
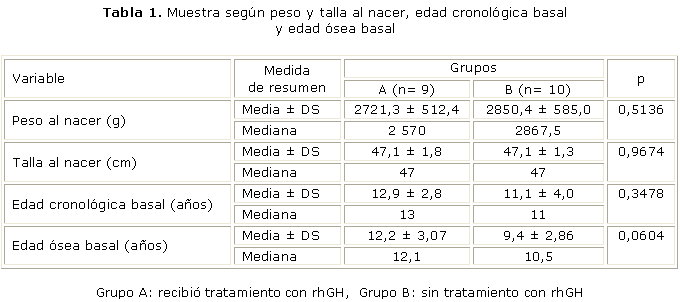
Se estudiaron 19 pacientes con el diagnóstico clínico y cromosómico de ST, que cumplían los criterios de inclusión establecidos. Se revisaron las historias clínicas de las pacientes y se formaron los 2 grupos. Se realizó una descripción de las características de las pacientes que demostró que ambos grupos son comparables, sin diferencias significativas en las variables (tabla 1), peso al nacer (p= 0,5136), talla al nacer (p= 0,9674), edad cronológica inicial (p= 0,3478), edad ósea inicial (p= 0,0604), función tiroidea (p= 0,9568), déficit de GH (p= 0,4371) y diagnóstico cromosómico (p= 0,5150). El grupo A mostró al inicio del estudio una edad cronológica media de 12,9 ± 2,8 años, y el grupo B de 11,1 ± 4,0 años. La edad ósea basal en el grupo A fue de 12,2 ± 3,0 años, y en el grupo B de 9,4 ± 2,8 años, sin diferencias significativas.
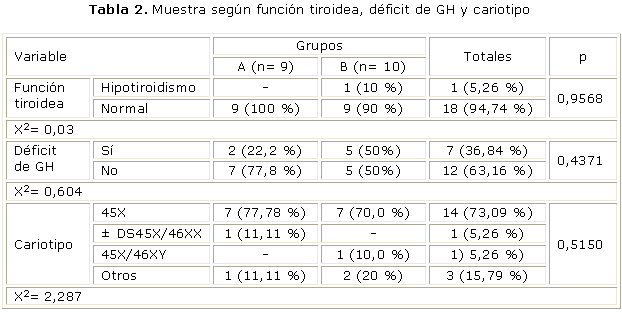
En la tabla 2 se observa la distribución de la muestra según función tiroidea, así como la interpretación de la prueba de clonidina y prueba de sensibilidad a la insulina, y la distribución de los sujetos según fórmula cromosómica. El progreso de la talla en las niñas con ST tratadas con rhGH se muestra en la tabla 3, y en la tabla 4 se detalla el comportamiento de la velocidad de crecimiento (Vc) en ambos grupos.

La comparación de la Vc entre el grupo A y B mostró los resultados siguientes: no se observó diferencia significativa en la Vc media basal (p= 0,3272), concluyendo el estudio con diferencias significativas en la Vc media al año (p= 0,0032) entre ambos grupos. La Td media del grupo A (160,0 ± 9,0 cm) y del grupo B (159,0 ± 12,0 cm) se ubican entre el percentil 50 y 75. Los 2 grupos según su talla media (basal y al año), y edad cronológica (basal y al año), se encuentran por debajo del percentil 3, pero el grupo A sigue una curva de crecimiento paralela a dicho percentil, mientras que el grupo B se separa progresivamente.
En la tabla 5 se muestra el peso basal y al año en ambos grupos. El grupo A presentó un peso basal medio de 34,0 ± 6,8 kg, y al año de 38,5 ± 6,9 kg, y teniendo en cuenta su relación peso para la edad se localizó en el 25 percentil al inicio y final del estudio. En el grupo B el peso basal medio fue de 28,0 ± 7,8 kg y al año de 29,7 ± 8,0 kg, por lo que se ubicó inicialmente, según la relación anterior, en el 25 percentil, para después de un año de estudio descender a un 10-25 percentil.

Teniendo en cuenta la relación peso para la talla, el grupo A y B al inicio del estudio se localizaron entre los percentiles 90-97, después de un año el grupo A mantiene la misma ubicación, mientras que el grupo B asciende para localizarse por encima del 97 percentil. Siete de las pacientes recibieron tratamiento con estrógeno con el fin de inducir la pubertad, 4 pertenecientes al grupo A y 3 al grupo B, con una edad cronológica media de 13,4 ± 0,5 años, y con una dosis media de 30 µ/d (anticonceptivos orales).
DISCUSIÓN
El hallazgo físico más constante en el ST es, sin duda, la baja talla, la cual se presenta en el 98 % de las pacientes.28,29 Los primeros intentos de tratamiento con GH en niñas con ST, realizados por Tanner y otros, en 1971, fueron poco satisfactorios; sin embargo, estos fracasos se atribuyeron a la edad de inicio del tratamiento, así como a la dosis y a la frecuencia de este. Posterior a esto, la mayoría de los estudios han mostrado la efectividad del tratamiento con GH biosintética, sola o combinada con oxandrolona.29
En nuestro estudio el grupo A mostró al inicio una edad cronológica media de 12,9 ± 2,8 años, y el grupo B de 11,1 ± 4,0 años (p= 0,3478). La edad ósea basal en el grupo A fue de 12,2 ± 3,0 años, y en el grupo B de 9,4 ± 2,8 años, sin diferencias significativas (p= 0,0604). Se registran otros estudios iniciados a una edad cronológica media de 12,9 años, con una ganancia neta de talla de 6,4 ± 4,9 cm.19 No se observaron los mismos resultados en la investigación de Van de Broeck, en el que, con igual edad cronológica media de inicio, se obtuvo una ganancia neta de talla de 2,9 ± 3,8 cm.19
Con una frecuencia mayor a la población general, las pacientes con ST padecen algunas enfermedades, en especial las autoinmunes, como la tiroiditis de Hashimoto y el hipotiroidismo, entre otras. Se conoce que del 25 al 30 % de las mujeres con ST son hipotiroideas, en comparación con el 1,5 % de las mujeres adultas de la población normal;19,28 sin embargo, la incidencia de la enfermedad de Graves no está aumentada.30-33 Otros reportes internacionales informan que del 10 al 30 % de las niñas con ST padecen hipotiroidismo primario debido usualmente a una tiroiditis linfocítica crónica.34
Encontramos en el grupo B una paciente con hipotiroidismo compensado, para un 10,00 %, que representó el 5,26 % del total de la muestra. El resto presentó una función normal para un 90,00%. Las 9 pacientes del grupo A mostraron una función tiroidea normal, para un 100 %. Radetti y otros, por su parte, estudiaron 478 mujeres con ST con una edad media de 15 años. El 22,5 % presentó anticuerpo antitiroideo positivo, de este último grupo el 27 % sufría de hipotiroidismo y el 3 % de tirotoxicosis. Las pacientes hipotiroideas representaron el 6 % del total de la muestra, lo cual se corresponde con nuestros resultados.30,35
Las pruebas de estimulación para valorar la secreción de GH en las niñas con ST suelen ser normales entres los 8 y 12 años de edad, a partir de esa edad existe una disminución, lo que es corroborado por Ranke y otros, que encontraron valores normales de GH hasta los 9 años de edad. Posteriormente, los niveles disminuyen significativamente hasta observarse una deficiencia relativa de GH durante los años correspondientes a la pubertad.29
En nuestra investigación se observaron 12 niñas sin déficit de GH, para un 63,16 % del total de la muestra, lo cual no arrojó diferencias significativas entre ambos grupos (p= 0,4371). Resultados parecidos mostraron Schmitt y otros,29 que observaron que el 31,5 % de las niñas con ST presentaba déficit de GH, distribuido de la forma siguiente: el 22 % de las pacientes estudiadas tuvieron niveles subnormales de GH en las pruebas de estimulación (picos máximos menores de 11 ng/mL) y 9,5 % presentó pruebas patológicas (picos máximos menores de 7 ng/mL). Otra investigación con resultados muy parecidos fue la realizada por el grupo italiano de estudio de ST, la cual incluyó 301 pacientes, y comprobaron que el 36,2 % tuvo una reducción en la reserva de GH en 2 pruebas de estimulación (GH< 10 ng/L) y el 61,8 % una disminución de la media de secreción espontánea nocturna (GH< 3,3 ng/L).11
La mayoría de las pacientes estudiadas mostraron una fórmula cromosómica 45X, para un 73,69 % del total de la muestra, los cariotipos 45X/46XX y 45X/46XY representaron el 5,26 %, mientras que 15,79 % se agrupó en otras fórmulas cromosómicas. No se mostraron diferencias significativas (p= 0,5150) entre ambos grupos con relación al diagnóstico cromosómico.
Algunas diferencias se muestran en un estudio realizado en el Hospital Middlesex y Oxford Radeliffe en el Reino Unido, donde se analizó el diagnóstico de 196 mujeres con ST, que mostró los resultados siguientes: el 48 % de la muestra presentó una fórmula cromosómica 45X, el 11 % correspondió a las pacientes con 45X/46XX, el 6 % coincidió con el cariotipo 45X/46XY, y el 35 % restante se distribuyó en otras fórmulas cromosómicas.30
En los estudios internacionales se reportan incrementos de talla final de 3,0 a 9,2 cm en comparación a la talla proyectada.34-37 Haeusler y otros estudiaron a 20 pacientes con ST, que llevaron tratamiento con GH inicialmente y añadieron tras 2 años oxandrolona hasta la talla final, con una edad cronológica media de inicio de tratamiento de 11,8 años, y se obtuvieron buenos resultados, avalados por una ganancia neta de talla de 9,3 cm.38
Buenos resultados se observaron con relación a la talla en las niñas con ST tratadas con rhGH, donde el grupo A inició el estudio con una talla basal media de 131,7 ± 7,5 cm, para mostrar una talla media después de un año de tratamiento de 137,9 ± 7,1 cm, que arrojó una diferencia significativa de p= 0,007. Sin embargo el grupo B presentó una talla basal media de 120,7 ± 15,1 cm y una talla media al año del estudio de 123,0 ± 14,0 cm. No se muestran diferencias significativas cuando comparamos la talla media basal entre ambos grupos (p= 0,050), y sí al comparar la talla media al año (p= 0,0071).
En nuestra investigación se observaron resultados positivos con relación a la Vc en el grupo A, que presentó una Vc media basal de 3,3 ± 1,5 cm/año, para mostrar después de un año de tratamiento una Vc media de 6,2 ± 2,3 cm/año, con una diferencia significativa de p= 0,0209. Resultados diferentes se observaron en el grupo B, con una Vc media al inicio del estudio de 2,4 ± 0,8 cm/año, y al terminar este, una Vc media de 2,2 ± 1,3 cm/año, sin arrojar diferencias significativas (p= 0,4008). No se observó diferencia significativa en la Vc media basal (p= 0,3272), y concluye el estudio con diferencias significativas en la Vc media al año (p= 0,0032) entre ambos grupos. Estos resultados son comparables con los obtenidos por Escamilla y otros,12 en un grupo de niñas con ST, a las cuales se les administró hGH, que mostró como resultados un incremento de la Vc de 3,8 a 7,5 cm/año.
Similares resultados, durante el primer año, presentó la experiencia belga12,19 del uso de hGH a dosis farmacológicas (0,95 ± 0,13 UI/kg/semana) en 49 niñas con ST, con un incremento de la Vc basal de 4,0 cm/año a 7,4, 5,8, 5,0 y 3,7 cm/año, durante 4 años consecutivos de tratamiento. En EUA,21 en 1983, se estudiaron 70 niñas con ST entre 4,7 y 12,4 años de edad, las cuales fueron tratadas por un período de 12 a 20 meses con hGH un primer grupo, y el segundo con hGH y oxandrolona. La Vc basal en el primer grupo fue de 4,5 cm/año, con un incremento de 6,6 cm/año en el primer año y en los 10 meses siguientes de tratamiento. En el estudio multicéntrico español32 se obtuvieron resultados parecidos en el grupo 1, tratado con rhGH exclusivamente, con una Vc media de 7,37 cm a los 12 meses del estudio, con relación a una Vc basal de 3,49 cm. Fernández por su parte, encontró un incremento de la Vc, en el grupo tratado con hGH sola, de 4,1 a 7,6 cm durante el primer año de tratamiento.29
La obesidad se observa con mayor frecuencia y particularmente durante la adolescencia en las niñas con ST. En nuestra experiencia ambos grupos iniciaron el estudio en el percentil 25 del peso para la edad, mientras que al finalizar este, el grupo B se encontraba entre los percentiles 10 y 25. El grupo A se mantuvo en el percentil de inicio. Se valoró la relación peso/talla, indicador que se tuvo en cuenta para definir la malnutrición por exceso en estas pacientes por presentar una baja talla, y se puede decir que los grupos A y B presentaron un sobrepeso al inicio del estudio (percentil 90-97). Después de un año, el grupo que recibió tratamiento mantuvo la misma condición, mientras que el grupo B sobrepasó el 97 percentil, que se considera como obesidad. El hecho de que el grupo A mantenga el mismo percentil al inicio y final del estudio, y no descienda o ascienda en una desviación estándar como el grupo B, según la relación peso para edad y peso para talla respectivamente, se debe al uso del tratamiento con rhGH, aunque en la relación peso para talla hay que tener en cuenta el cumplimiento de las orientaciones médicas por el paciente para evitar o tratar la desnutrición por exceso.
Se indujo la pubertad en 7 pacientes, que recibieron estrógeno a una dosis media de 30 mcg/día, 4 pertenecientes al grupo A y 3 al grupo B, con una edad cronológica media de 13,4 ± 0,5 años. La pubertad fue inducida a edades muy similares en las pacientes estudiadas por Massa G y otros,39 con buenos resultados sobre la talla final, según muestra en su investigación representada por 186 niñas con ST tratadas con GH (dosis media de 33 mg/kg/semana a una edad media inicial de 11,6 años) hasta la talla final. Se le indujo la pubertad a 148 pacientes (dosis media inicial de etinilestradiol de 66 ng/kg/día) a una edad media de 14,5 años, con una talla media al inicio de la pubertad de 143,3 cm, y alcanzaron una talla final media de 151,8 cm (talla diana 162,1 cm).
Otros autores coinciden en inducir la pubertad a una edad posterior a los 14 años, como Chernouse SD y otros,40 que formaron 2 grupos de pacientes con ST: un primer grupo (n= 26) en el que se inicia la terapia sustitutiva con estrógeno a los 12 años de edad (premarin a una dosis de 0,3 mg/día los primeros 6 meses, aumentando a 0,625 mg/día los siguientes 6 meses, y después de un año se agregó acetato de medroxiprogesterona a 10 mg/día por 10 días, con frecuencia mensual); y un segundo grupo (n= 29), que inició el tratamiento a los 15 años. Ambos grupos recibieron tratamiento con rhGH a una dosis de 0,375 mg/kg/semana, inyectada en cualquier momento del día, 3 veces a la semana, a una edad inicial de 9,5 años, y se concluyó que el progreso de la edad ósea fue significativamente mayor en el primer grupo con relación al segundo (p= 0,004). Durante el primer año de estudio la tasa de crecimiento fue mayor en el grupo que inició la terapia a los 12 años, pero posteriormente, durante el segundo año de administración de premarin, la tasa de crecimiento en el grupo que inició tratamiento a la edad de 12 años disminuyó, mientras que se mantuvo en el segundo grupo. Terminó la investigación con una talla final 3,4 cm mayor en el grupo que inició el tratamiento a los 15 años (talla final 150,4 vs. 147,0 cm).
Otros autores, como Hochberg Z y otros,40,41 demuestran en su estudio que la mayoría de las pacientes con ST expresan el deseo de iniciar la pubertad a la misma edad que sus compañeras de aula, y agregan además, que si se induce la pubertad a edad fisiológica comenzando el primer año con una dosis de 50 ng/kg/día de etinilestradiol, aumentando a 100 ng/día en el segundo año, y a 200 ng/día en el tercer año, no se influye sobre la talla final. Existen 3 trabajos muy ilustrativos que demuestran el efecto negativo que ejerce el inicio precoz del tratamiento con estrógeno sobre la talla final. Attie y otros,19,42 Werther y otros,20,43 y el estudio sueco de Nilsson y otros,20,44 concluyen que con la administración de estrógeno a partir de los 13 años se logra una mejor talla final.
Se concluye señalando que el tratamiento con rhGH durante el primer año resultó efectivo, al acelerar significativamente la velocidad de crecimiento en las niñas con ST. La ganancia de peso corporal resultó adecuada durante el periodo de estudio, en el cual se logró mantener una valoración nutricional estable sin modificaciones en el canal percentilar. La inducción de la pubertad no cambió el pronóstico de la talla al final del estudio.
REFERENCIAS BIBLIOGRÁFICAS
1. Turner HH. A syndrome of infantilism, congenital webbed neck, and cubits valgus. Endocrinology. 1938;23:566-74.
2. Lyon AJ, Preece Ma, Grant DB. Growth curve for girls with Turner syndrome. Arch Dis Child. 1985;60:932-5
3. Padrón RS, Barón JA, Arce B. Correlación fenotipo-cariotipo en el síndrome de Turner. Rev Cub Med. 1981;20:62-7.
4. Cavallo L, Gurrado R. Endogenous growth hormone secretion does not correlate with growth in patients with Turner's syndrome. J Pediatr Endocrinol Metab. 1999;12:623-7.
5. Massa G, Vanderchueren-Lodewyckx M, Malvaux P. Linear growth in patients with Turner' syndrome: influence of spontaneous puberty and parental height. Eur J Pediar. 1990;149:246-50.
6. Page LA. Final heights in 45 X Turner syndrome with spontaneous sexual development. Review of European and Americans reports. J Pediatr Endocrinol. 1993;6:153-8.
7. Ogata T, Muroya K, Matsuo N. Structure function relation of the X chromosome in Turner Syndrome. En: Saenger P, Pasquino AM, eds. Optimising health care for Turner patients in the 21st century. Amsterdam: Elsevier; 2000. p. 9-18.
8. Ellison WJ, Wardak Z, Yonung MF, Gehron-Rebey P, Laig-Webster M, Chiong W. PHOG, a candidate gene for involvement in the short stature of Turner syndrome. Hum Mol Genet. 1997;6:1341-7.
9. Rao E, Weiss B, Fukami M, Rump A, Niesler B, Mertz A, et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet. 1997;16:54-63.
10. Kosho T, Muroya K, Nagai T, Fujimoto M, Yokoya S, Sakamoto H, et al. Skeletal features and growth patterns in 14 patients with haplo insufficiency of SHOX: implications for the development of Turner syndrome. J Clin Endocrinol Metab. 1999;84:4613-21.
11. Takano K, Ogawa M, Tanaka T, Tachibana K, Fujira K, Hisuka N. Clinical trials of GH treatment in patients with Turner's syndrome in Japan-a consideration of final height. J Endocrinol. 1997;137:138-45.
12. Nelly EK, Rosenfeld RG. Turner Syndrome. En: Lifshitz F. Pediatric Endocrinology. 3a ed. New York: Marcel Dekker; 1996. p. 267-80.
13. Ross JL, Long LM, Loriaux DL, Cutler GB, Growth hormone secretory dynamics in Turner syndrome. J Pediatr. 1985;106:202-6.
14. Rongen-Westerlanken C, van Es A, Wit J-M. Growth hormone therapy in Turner's syndrome. J Des Child. 1992;146:817-20.
15. Stahnke N, Stubbe P, Keller E. Recombinant human growth hormone and oxandrolone in treatment of short stature in girls with Turner syndrome. Horm Res. 1992;37(Suppl 2):37-46.
16. Rosenfield RG, Frane J, Attie KM. Six year results of a randomized prospective trial of human growth hormone and oxandrolone in Turner Syndrome. J Pedriatr. 1992;121:49-55.
17. Garcia RC, Martinez A, Helnrich JJ, Kelcenman A, Lejaraga N, Laspiur M. Growth in Argentinean girls with Turner syndrome. Ann Hum Biol. 1995;22:533-44.
18. Rochiccioh P, Battin J, Bertrand AM, Bost M, Cabrol S. Final height in Turner syndrome patients treated with growth hormone. Horm Res. 1995;44:172-6.
19. Ferrández A, Labarta JI, Calvo M, Mayayo E, Puga B, Cáncer E, et al. Síndrome de Turner. En: Pombo M. Tratado de Endocrinología Pediátrica. 3a ed, Madrid: McGraw-Hill Interamericana; 2002. p. 780-803.
20. Luzuriaga C. Tratamiento con hormona de crecimiento y aspectos básicos de su administración. En: Luzuriaga C. Crecimiento y hormona de crecimiento. Salamanca: Técnica de congresos TESITEX; 1994. p. 167-91.
21. Argente OJ, Carrascosa LA, Gracia BR. Pruebas Funcionales en Endocrinología Pediátrica y de la Adolescencia. En: Argente OJ, Carrascosa LA, Gracia BR, Rodríguez HF. Tratado de Endocrinología Pediátrica y de la Adolescencia. Madrid: EDITORES MÉDICOS, S.A; 1995. p. 1075-136.
22. Vicen CE. Método de exploración de la secreción hormonal adeno-hipofisaria. En: Pombo M. Tratado de Endocrinología Pediátrica. 3a ed, Madrid: McGraw-Hill Interamericana; 2002. p. 403-9.
23. Luzuriaga C. Valoración de la secreción de Hormona de Crecimiento y Somatomedina. Prueba analítica. En: Luzuriaga C. Crecimiento y Hormona de crecimiento, Salamanca: Técnica de congresos TESITEX. 1994. p. 115-40.
24. Bona X, Luzuriaga C. Materiales y métodos para la medida y valoración del crecimiento. En: Luzuriaga C. Crecimiento y Hormona de crecimiento. Salamanca: Técnica de congresos TESITEX; 1994. p. 219-36.
25. Fernández RM. Patrón de crecimiento humano y su evaluación. En: Pombo M. Tratado de Endocrinología Pediátrica. 3a. ed. Madrid: McGraw-Hill Interamericana; 2002. p. 244-74.
26. Garagori JM, Moreno LA. Talla baja familiar y retraso constitucional del crecimiento y el desarrollo. En: Argente OJ, Carrascosa LA, Gracia BR, Rodríguez HF. Tratado de Endocrinología Pediátrica y de la Adolescencia. Madrid: EDITORES MÉDICOS, S.A; 1995. p. 141-53.
27. Pozo J, Argente J. Crecimiento: Valoración auxológica. En: Argente J, Carrascosa A, Gracia R, Rodríguez F. Tratado de Endocrinología Pediátrica y de la Adolescencia. 2a ed. Barcelona: Doyma; 2000. p. 177-200.
28. Rodríguez HF, Ballester F. Disgenesias Gonadales. En: Argente OJ, Carrascosa LA, Gracia BR, Rodríguez HF. Tratado de Endocrinología Pediátrica y de la Adolescencia. Madrid: EDITORES MÉDICOS, S.A; 1995. p. 141-53.
29. García NL, Luis DA, Lus SP. Uso de la hormona de crecimiento biosintética en el síndrome de Turner. En: Raúl CL, Luis DA. Fisiopatología de la pubertad y el embarazo/anticoncepción en la adolescencia. México: Dorantes Alvarez; 1997. p. 371-85.
30. Elsheikh M, Dunger DB, Conway GS, Wass JA. Turner syndrome in adulthood. Endocrinol rev. 2002;23:120-40.
31. Sybert VP. The adult patient with Turner syndrome. En: Albertsson WK, Ranke MB. Turner syndrome in a life span perspective: research and clinical aspects. Amsterdam: Elsevier; 1995. p. 205-18.
32. Weiss L. Additional evidence of gradual loss of germ cells in the pathogenesis of streak ovaries in Turner's syndrome. J Med Genet. 1971;8:540-4.
33. Elsheikh M, Wass JAH, Conway GS. Autoimmune thyroid disease in women with Turner's syndrome-the association with karyotype. Clin Endocrinol. 2001;55:223-6.
34. Stephure KD. Turner Syndrome. International Symposium on A Current of Pediatric Endocrinology. 1997;105-10.
35. Radetti G, Mazzanti L, Paganini C, Bernasconi S, Russo G, Rigon F, et al. Frequency, clinical and laboratory features of thyroiditis in girls with Turner's syndrome. The Italian Study Group for Turner 's syndrome. Acta Paediatr. 1995;84:909-12.
36. Rosenfeld RG and the Genentech National Cooperative Study Group. Growth hormone therapy in Turner's syndrome: an update on final height. Acta Paediatr Suppl. 1992;383:2-3.
37. Van den Broeck J, Massa GG, Attanasio A, Matranga A, Chaussain JL, Price DA, et al. Final height after long-term growth hormone treatment in Turner syndrome. J Pediat. 1995;127:729-35.
38. Haeusler G, Schmitt K, Blümel P, Plöchl E, Waldhör TH, Frisch H. Growth hormone in combination with anabolic steroids in patients with Turner syndrome: effect on bone maturation and final height. Acta Peadiatr. 1996;85:1408-14.
39. Massa G, Heinrichs C, Verlinde S, Thomas M, Bourguignon JP, Croen M, et al. Late or delayed induced or spontaneous puberty in girls with Turner syndrome treated with growth hormone does not affect final height. International Growth Monitor Rev. 2004;14:15-7.
40. Chernausek SD, Attie KM, Cara JF, Rosenfeld RG, Frane J. Growth hormone therapy of Turner syndrome: the impact of age of estrogen replacement on height. International Growth Rev Monitor. 2002;11:20-2.
41. Hochberg Z, Zadik Z. Final height in young women with Turner syndrome after GH therapy: an open controlled study. Eur Endocrinol. 1999;141:218-24.
42. Atti KM, Chernausek S, Frane J. Growth hormone use in Turner syndrome: a preliminary report on the effect of early versus delayed estrogen. En: Albertsson WK, Ranke MB. Turner syndrome in a life-span perspective. Research and clinical aspects. Amsterdam: Elsevier Science; 1995. p. 175-81.
43. Werther G, Diestsch S. Multicentre trial of synthetic growth hormone and low dose estrogen in Turner syndrome: analysis of final height. En: Albertsson WK, Ranke MB. Turner syndrome in a life-span perspective. Research and clinical aspects. Amsterdam: Elsevier Science; 1995. p. 105-12.
44. Nilsson OK, Albersson WK, Alm J. Improved final height in girls with Turner's syndrome treated with growth hormone and oxandrolone. J Clin Endocrinol Metab. 1996;81:635-40.
Recibido: 28 de noviembre de 2012.
Aprobado: 14 de enero de 2013.
Tania Mayvel Espinosa Reyes. Instituto Nacional de Endocrinología. Calle Zapata y D, Vedado, municipio Plaza de la Revolución. La Habana, Cuba. Correo electrónico: tania.espinosa@infomed.sld.cu

{kind=link}
{kind=link}
{kind=link}