










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Cubana de Endocrinología
versão On-line ISSN 1561-2953
Rev Cubana Endocrinol vol.24 no.3 Ciudad de la Habana set.-dez. 2013
PRESENTACIÓN DE CASO
Carcinoma adrenal de rápida evolución
Rapidly evolving adrenal carcinoma
Dra. Teresa Margarita González Calero, Dra. Ana Ibis Conesa González, Lic. Julio César Hernández Martínez, Dra. Lisett Leal Curi
Centro de Atención al Diabético. Instituto Nacional de Endocrinología (INEN). La Habana, Cuba.
RESUMEN
Introducción: el carcinoma suprarrenal primario es un tumor poco frecuente, altamente agresivo, de crecimiento rápido, con mayor incidencia entre los 40 y 60 años de edad. Los carcinomas funcionantes representan hasta un 79 % de los tumores corticales, más frecuentes en el sexo femenino, y de estos el 50 % se manifiestan clínicamente como un síndrome de Cushing. La extensión del tumor a estructuras vecinas es común y empeora el pronóstico. La supervivencia media es de 2 años desde el diagnóstico, en particular, cuando existen metástasis en hígado y pulmón.
Objetivo: describir las características clínicas, los procederes diagnósticos y terapéuticos de una paciente con carcinoma adrenal de rápida evolución.
Presentación del caso: paciente LRS, femenina, blanca, de 49 años de edad, con antecedentes de hipertensión arterial y diabetes mellitus tipo 2, que asiste a consulta por descontrol metabólico y de la hipertensión arterial. Al examen físico, se constatan signos sugestivos de hipercortisolismo, sintomatología que a los 2 meses se acentuó notablemente. Se realizaron estudios basales, dinámicos, imagenológicos y anatomopatológicos, que corroboraron el diagnóstico presuntivo. Se realiza adrenalectomía izquierda con adenectomía regional, y se confirma por anatomía patológica el diagnóstico de carcinoma suprarrenal izquierdo. A los pocos meses de la intervención la paciente fallece con metástasis óseas en columna vertebral.
Conclusiones: el reconocimiento temprano de los síntomas y signos de hiperfunción adrenal es muy importante para el diagnóstico y tratamiento oportuno del carcinoma adrenal.
Palabras clave: carcinoma adrenal, síndrome de Cushing, neoplasia, hipercortisolismo endógeno.
ABSTRACT
Introduction: primary suprarrenal carcinoma is a highly aggressive rare carcinoma of rapid growth, with greater incidence in 40-60 years age group. The functioning carcinomas represent up to 79 % of cortical tumors that are more frequent in females, and 50 % of them clinically manifests as Cushing syndrome. The extension of the tumor to neighboring structures is common and worsens the prognosis. Mean survival rate is 2 years from the time of diagnosis, particularly when there are liver and lung metastases.
Objective: to describe the clinical characteristics, the diagnostic and therapeutic procedures in a female patient with rapidly evolving adrenal carcinoma.
Case presentation: a patient LRS, female, Caucasian, 49 years-old, with a history of blood hypertension and type 2 diabetes mellitus, who went to the hospital because of lack of metabolic control and blood hypertension. On physical examination, there were observed some signs suggestive of hypercortisolism, symptomatology that became notably acute two months later. Basal, dynamic, imaging and anatomopathological studies were made to corroborate the presumptive diagnosis. Left adrenalectomy with regional adenectomy was performed, and the diagnosis of left suprarenal carcinoma was confirmed through pathological anatomy. Few months later, the patient died from osseous metastasis in her spinal cord.
Conclusions: early recognition of symptoms and signs of adrenal hyperfunction is very important for diagnosis and timely treatment of adrenal carcinoma.
Keywords: adrenal carcinoma, Cushing syndrome, neoplasia, endogenous hypercortisolism.
INTRODUCCIÓN
El carcinoma suprarrenal primario es un tumor poco frecuente, de baja incidencia (1-2 casos por 1 000 000 habitantes/año), altamente agresivo y de mal pronóstico, que ocasiona entre 0,05 y 0,2 % de las muertes por cáncer, con mayor incidencia entre los 40 y 60 años de edad. En niños es infrecuente, y solo representa el 6 % de los tumores adrenales en la infancia. La supervivencia es el doble que en adultos, y la mayoría son hormonalmente activos.1-4 Se pueden dividir en neoplasias funcionantes y no funcionantes. Las funcionantes, representan hasta 79 % de los tumores corticales, son más frecuentes en el sexo femenino, y de estos, el 50 % se observa clínicamente como un síndrome de Cushing.4-6 Cerca del 40 % de los carcinomas adrenocorticales no segregan ninguna hormona ni provocan síntomas específicos, se descubren accidentalmente, o como parte de una evaluación periódica. Dada su localización anatómica suelen alcanzar gran tamaño, por ser de diagnóstico más tardío que los que producen síntomas hormonales.6-9
La etiología es desconocida. Recientemente se han utilizado para este fin marcadores moleculares: factores de crecimiento 1 y 2 semejantes a la insulina (IGF-1) e (IGF-2), y genéticos: estudio de genes heterocigotos 11P, 13Q o 17P, involucrados en este tipo de entidad.1,9,10
El carcinoma cortical puede originarse de "células adrenales en reposo", formas de tejido adrenocortical accesorio o aberrante. Se han localizado en el ligamento ancho, testículo (especialmente en niños), y en el polo superior del riñón. Más raramente se han localizado en posición intracraneal, el hígado o el pulmón.1,10,11 Se describen como factores de riesgo: el sexo femenino, la edad (menor de 5 años, y entre 40 y 50 años) y la herencia.1,12,13 La clínica de los carcinomas adrenales debe ser considerada en 3 categorías:1 9,14-17
- Síntomas debidos a la propia masa adrenal: síntomas gastrointestinales, astenia, fatiga, pérdida de peso, febrícula y dolor abdominal de inicio brusco, masa palpable (30-40 %).
- Manifestaciones secundarias a la diseminación regional y/o a distancia: metástasis pulmonares (60 %), hepáticas (50 %), a ganglios linfáticos (48 %) y óseas (23 %); o localmente, a riñones, vena suprarrenal, vena cava, ganglios regionales y periaórticos.
- Síntomas endocrinos de hiperfunción: dadas por manifestaciones clínicas del síndrome de Cushing (distribución centrípeta de la grasa, cifosis dorsal, fascie de luna llena con rubicundez facial, acné, hirsutismo, estrías violáceas en abdomen y glúteos); así como también virilización en las mujeres, feminización en los hombres y patrones mixtos.
El diagnóstico clínico se complementa con:
1. Estudios de laboratorio: la evaluación debe incluir química sanguínea, electrolitos séricos, pruebas de coagulación, función renal y hepática, marcadores tumorales (antígeno carcinoembrionario CA 19,9 y 125), fosfatasa alcalina y DHL, así como pruebas hormonales especificas (testosterona libre, dihidroepiandrosterona sulfato [DHEA-s], cortisol sérico, adrenocorticotropina [ACTH], cortisoluria 24 horas y test de supresión con 8 mg de dexametasona).1,9,12,13,18
2. Estudios imagenológicos como:1,12,18-21
- Tomografía axial computarizada (TAC): con o sin medio de contraste se han utilizado para diferenciar masas benignas o malignas suprarrenales basadas en su contenido lipídico.
- Ultrasonido: las lesiones malignas varían en textura y ecogenicidad y presentan apariencia hemorrágica y/o calcificaciones.
- Resonancia magnética: los tumores aparecen como masas hipointensas en comparación a T-1 e hiperintensas T-2 con respecto al hígado. Muestra la invasión o desplazamiento de órganos.
- Estudios histológicos: se toman en cuenta 9 criterios histológicos, y el principal se define como un alto grado de mitosis (más de 20 por 10 campos de poder).18,19
El estadiaje del carcinoma suprarrenal se realiza sobre la base del tamaño tumoral, la extensión regional y las estructuras relacionadas, usando las tablas de Mc Farlane (modificadas de las tablas de Sullivan).12
Se estima que el tratamiento quirúrgico en etapas tempranas es el mejor método de supervivencia para el paciente, si se asocia con terapia adyuvante. Se sugiere adrenalectomía en lesiones sólidas entre 3-4 cm, especialmente en pacientes jóvenes, y linfadenectomía periaorticocava, apoyado con quimioterapia.18,19-23 Es controversial la adrenalectomía laparoscópica por el alto riesgo de recurrencia.24-26
La quimioterapia se mantiene como una ayuda temporal en tumores de estadio avanzado, y de manera terapéutica para estadios iniciales. Algunos autores recomiendan la administración posoperatoria de forma precoz de mitotane, para control del volumen tumoral y disminuir los síntomas secundarios a la producción hormonal.9,12,18,19,27
La extensión del tumor a estructuras vecinas es frecuente y empeora el pronóstico, con rangos de supervivencia entre un 12,5 % en pacientes con enfermedad regional y 5,5 % con enfermedad a distancia, aunque la combinación de quimioterapia y tratamiento quirúrgico puede extender significativamente la expectativa de vida en pacientes en estadio I, II y III. Obviamente, el diagnóstico temprano es la mejor arma con que cuenta el clínico, al observar las manifestaciones de exceso hormonal, en contraste con el sombrío cuadro de los estadios avanzados.1,9,12,19,28
PRESENTACIÓN DEL CASO
Paciente LRS, femenina, blanca y de 49 años de edad al momento del diagnóstico. Como antecedentes patológicos personales se señala hipertensión arterial (HTA) tratada con atenolol 100 mg/día y clortalidona 25 mg/día, y diabetes mellitus tipo 2 (DM 2) diagnosticada en su segunda gestación, con 16 años de evolución y tratamiento con dieta y glibenclamida a dosis máxima (20 mg al día).
Se decide su ingreso en el Centro de Atención al Diabético del Instituto Nacional de Endocrinología (INEN) por descontrol metabólico y cifras elevadas de tensión arterial. Durante el ingreso se asocia metformina (850 mg, 2 tabletas al día para el tratamiento antihiperglucemiante) y se cambia el tratamiento antihipertensivo a enalapril (40 mg/día) asociado al diurético, con lo cual se logró buen control glucémico y de las cifras de tensión arterial. Durante el ingreso, al examen físico, se constatan signos sugerentes de hipercortisolismo: fascie de luna llena, piel fina y rubicunda, giba de búfalo y abdomen globuloso, por lo que se indican estudios para confirmar esta sospecha clínica.
En 2 meses acude a la consulta con clínica más acusada: vello terminal en mentón y región del bigote, astenia marcada, piel fina, con mayor rubicundez y lesiones micóticas circunscritas, hematomas ante traumas mínimos, onicomicosis generalizada en ambos pies, así como marcado descontrol glucémico y de la tensión arterial. El resultado del cortisol posinhibición con 2 mg de dexametasona resulta elevado, lo que confirma la sospecha clínica. Con estos elementos se decide su ingreso en la sala de Endocrinología del INEN, para completar estudios bioquímicos, hormonales e imagenológicos, y definir conducta.
Datos positivos al examen físico:
Examen físico general: cara redonda y rubicunda (fascie de luna llena), distribución centrípeta de las grasas y giba de búfalo.
Examen físico por aparatos y sistemas:
Piel: fina y rubicunda en cara, cuello y región supraclavicular, con lesiones de acné en la cara y el tronco. Se observa, además, presencia de vello terminal distribuida en diferentes regiones del cuerpo (cuadro 1), así como estrías nacaradas de 1 cm de grosor localizadas en hemiabdomen inferior. Igualmente presentaba lesiones hiperpigmentadas, aterciopeladas en región posterior del cuello (acantosis nigricans).
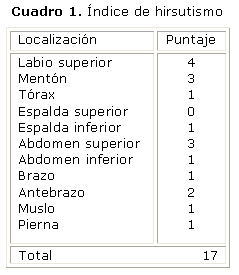
Faneras: uñas de ambos pies deslustradas, engrosadas, con pérdida en su continuidad.
Tejido celular subcutáneo (TCS): edemas en ambos miembros inferiores, que deja ligero godet.
Sistema vascular periférico: várices en ambos miembros inferiores.
Mensuraciones: (cuadro 2).

Durante el ingreso en el INEN se realiza:
- Cortisol basal: 516 nmol/L.
- Cortisol posinhibición rápida con 2 mg de dexametasona (Dxm): 812 nmol/L (no se inhibe).
- Cortisol posinhibición con 8 mg de Dxm: 624 nmol/L (no se inhibe).
- DHEA-s: 1,97 nmol/L.
- Ultrasonido abdominal: imagen compleja con calcificaciones en territorio suprarrenal izquierdo, de 50 x 45 x 44 mm, con imagen sugestiva de adenopatía, que mide 30 x 19 mm.
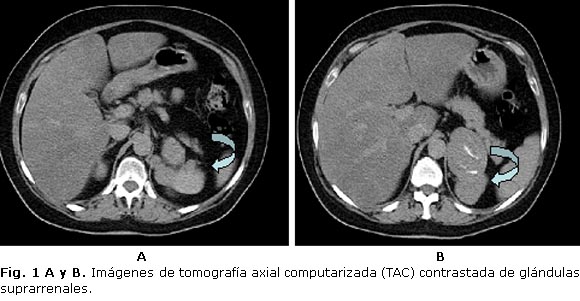
- TAC contrastada de suprarrenal: imagen hiperdensa tumoral en suprarrenal izquierda con calcificaciones de 49 mm de diámetro (figura 1 A y B).
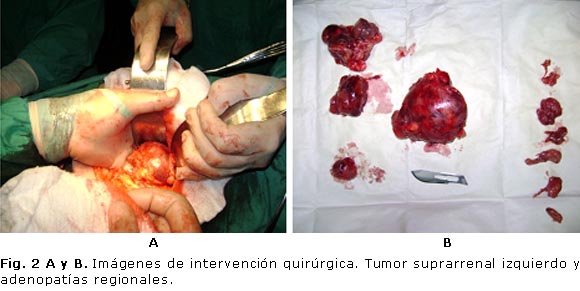
Confirmado el diagnóstico, se decide tratamiento quirúrgico por vía convencional, y se realiza adrenalectomía izquierda con adenectomía regional (figura 2 A y B). Se confirma el diagnóstico por los resultados de anatomía patológica.
Informe de anatomía patológica:
Descripción microscópica: glándula suprarrenal izquierda con múltiples fragmentos de tejido, que en conjunto, miden 11 x 6 x 5 cm, de consistencia fibroelástica, con áreas renitentes que muestran color pardo grisáceo, con área gris pálida al corte. Muestra una superficie con tendencia a la lobulación, con tejido central reblandecido, acompañado de zonas dispersas pequeñas, amarillas y firmes.
Descripción microscópica: carcinoma corticosuprarrenal izquierdo.
Al egreso, se decide seguimiento conjunto con el Instituto Nacional de Oncología (INOR), donde llevó tratamiento con quimioterapia y radioterapia.
En 4 meses la paciente refiere síntomas dolorosos en la columna, que se hacen más intensos progresivamente, por lo cual se realiza TAC que confirma la presencia de múltiples metástasis óseas. Fallece poco tiempo después.
El reconocimiento temprano de los síntomas y signos de hiperfunción adrenal y la sospecha de carcinoma adrenal, es importante para el diagnóstico temprano y tratamiento oportuno de esta entidad con tal mal pronóstico.
REFERENCIAS BIBLIOGRÁFICAS
1. Stewart PM, Krone NP. The Adrenal Cortex. En: Melmed S, Polonsky KS, Larsen PR, Kronenberg HM (eds). Williams Textbook of Endocrinology. 12th. ed. Philadelphia: Elsevier Saunder; 2011. p. 479-534.
2. Lal G, O'Dorisio T, Mc Dougall R, Weigel RJ. Cancer of the endocrine system. In: Abeloff MD, Armitage JO, Lichter AS, Niederhuber JE, Kastan MB, McKenna WG. Clinical Oncology. Philadelphia, PA: Elsevier; 2008. p. 1271-303.
3. Song JH, Chaudhry FS, Mayo-Smith WW. The incidental adrenal mass on CT: prevalence of adrenal disease in 1,049 consecutive adrenal masses in patients with no known malignancy. AJR Am J Roentgenol. 2008;190:1163-8.
4. Lacroix A. Approach to the Patient with Adrenocortical Carcinoma. J Clin Endocrinol Metab. 2010;95(11):4812-22.
5. Alonso Z, Vargas A. Neoplasias cortico-suprarrenales funcionantes. Reporte de un caso y revisión. Revista Médica de Costa Rica y Centroamérica LXII. 2005;573:179-83.
6. Linda NG, Libertino JM. Adrenocortical carcinoma: diagnosis, evaluation and treatment. J Urol. 2003;169(1):5-11.
7. Menéndez C, Casal F, Prieto J, Cacho L, Tuson C. Carcinoma suprarrenal. Análisis retrospectivo de cinco casos. An Med Interna. 2006;23(11):533-6.
8. Muro GE, Losada JL, Martín A, Pérez IR. Carcinoma suprarrenal gigante. Presentación de un caso. Arch Esp Urol. 2009;62(2):12-4.
9. Abiven G, Coste J, Groussin L. Clinical and biological features in the prognosis of adrenocortical cancer: poor outcome of cortisol-secreting tumors in a series of 202 consecutive patients. J Clin Endo & Metab. 2006;91:2650-5.
10. Kasperlik-Zaluska A, Otto M, Cichocki A, Roslonowska E, Slowiñska-Srzednicka J, Jeske W, et al. Incidentally Discovered Adrenal Tumors: A Lesson from Observation of 1444 Patients. Horm Metab Res. 2008;40(5):338-41.
11. William F, Young Jr. The Incidentally Discovered Adrenal Mass. N Engl J Med. 2007;356:601-10.
12. American Joint Committee on Cancer. Adrenal. In: AJCC Cancer Staging Manual. 7th. ed. New York: Springer; 2010. p. 515-7.
13. Fraker DL. Adrenal Tumors. In: DeVita VT, Hellman S, Rosenberg SA, eds. Cancer: Principles and Practice of Oncology. 8th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008. p. 1690-702.
14. Rodríguez-Hermosa JI, Roig J, Ortuno P, Quiles AM, Recasens M, Codina-Cazador A. Gran tumor suprarrenal. Rev Esp Enferm Dig. 2008;100(6):363-4.
15. Vera NV, AttanAsío RA, Chiesa LF, Elliff E, Menvielle S. Carcinoma adrenal secretor de estrógenos, a propósito de un caso. RAEM. 2009;46:84.
16. Mir M, Ruiz-Giménez N, Zapata C, Rodríguez B, Vílchez V, Suárez C. Carcinoma suprarrenal. Presentación de un caso y revisión de la literatura. Rev Clin Esp. 2009;209(3):150-2.
17. Mediavilla JD, López de la Torre M, Muñoz J, Sánchez MJ, Jaén F. Diversidad en la expresión clínica del carcinoma suprarrenal. Presentación de 7 casos. Endocrinología y Nutrición. 2004;51(8):82-6.
18. Allolio B, Hahner S, Weismann D. Management of adrenocortical carcinoma. Clin Endocrinol (Oxf). 2004;60:273-87.
19. Fassnacht M, Johanssen S, Quinkler M; German Adrenocortical Carcinoma Registry Group; European Network for the Study of Adrenal Tumors. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: proposal for a Revised TNM Classification. Cancer. 2009;115:243-50.
20. Pennanen M, Raade M, Louhimo J, Sane T, Heiskanen I, Arola J, et al. Adrenocortical tumours: high CT attenuation value correlates with eosinophilia but does not discriminate lipid-poor adenomas from malignancy J Clin Pathol. 2013;66(12):1076-80.
21. Sane T, Schalin-Jäntti C, Raade M. Is Biochemical Screening for Pheochromocytoma in Adrenal Incidentalomas Expressing Low Unenhanced Attenuation on Computed Tomography Necessary? J Clin Endocrinol Metab. 2012;97(6):2077-83.
22. Grubbs EG, Callender GG, Xing Y, Perrier ND, Evans DB, Phan AT, et al. Recurrence of Adrenal Cortical Carcinoma Following Resection: Surgery Alone Can Achieve Results Equal to Surgery Plus Mitotane. Ann Surg Oncol. 2010;17(1):263-70.
23. Wen TS, Cord S, Quan-Yang D. From incidentaloma to adrenocortical carcinoma: the surgical management of adrenal tumors. Journal of Surgical Oncology. 2005;89(3):186-92.
24. Kulis T, Knezevic N, Pekez M, Kastelan D, Grkovic M, Kastelan Z. Laparoscopic Adrenalectomy: Lessons Learned from 306 Cases. J Laparoendosc Adv Surg Tech A. 2012;22(1):22-6.
25. Zografos GN, Vasiliadis G, Farfaras AN, Aggeli C, Digalakis M. Laparoscopic surgery for malignant adrenal tumors. JSLS. 2009;13(2):196-202.
26. Cobb W, Kercher KW, Sing RF, Heniford B. Laparoscopic adrenalectomy for malignancy. The American Journal of Surgery. 2005;189(4):405-11.
27. Terzolo M, Angeli A, Fassnacht M. Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med. 2007;356:2372-80.
28. Choi YA, Kim CK, Park BK, Kim B. Evaluation of Adrenal Metastases from Renal Cell Carcinoma and Hepatocellular Carcinoma: uUse of Delayed Contrast-enhanced CT. Radiology. 2013;266(2):514-20.
Recibido: 2 de agosto de 2013.
Aprobado: 26 de noviembre de 2013.
Teresa Margarita González Calero. Centro de Atención al Diabético. Instituto Nacional de Endocrinología. Calle 17, esquina a D, Vedado, municipio Plaza. La Habana, Cuba. Correo electrónico: teregonzalez@infomed.sld.cu
