










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Endocrinología
versión On-line ISSN 1561-2953
Rev Cubana Endocrinol vol.24 no.3 Ciudad de la Habana sep.-dic. 2013
ENFOQUE ACTUAL
Interferón alfa y diabetes mellitus tipo 1
Interferon alpha and type 1 diabetes mellitus
Lic. Alieski Cruz Ramírez,I MSc. Lic. Yoima Rodríguez Marin,II Dr. C. Iraldo Bello RiveroI
ICentro de Ingeniería Genética y Biotecnología (CIGB). La Habana, Cuba.
IIInstituto Nacional de Endocrinología (INEN). La Habana, Cuba.
RESUMEN
La diabetes mellitus tipo 1 es una enfermedad autoinmune que se caracteriza por la destrucción de las células b pancreáticas debido a una respuesta autoinmune específica contra estas células, determinada por la interacción entre la susceptibilidad genética y factores ambientales no bien definidos. El interferón alfa juega un importante papel como mediador de la respuesta inmune innata y adaptativa del organismo, y se ha observado en estrecha relación con la infección por virus y la iniciación de la diabetes mellitus tipo 1. La aparición de autoanticuerpos naturales contra el interferón alfa ha sido objeto de atención por la comunidad científica internacional en las últimas décadas, y es un evento que se describe en aproximadamente el 10 % de los pacientes que presentan enfermedades de tipo autoinmune, entre las cuales se incluye la diabetes mellitus tipo 1. Sin embargo es muy limitada la información sobre su frecuencia, especificidad, inmunorreactividad y rol fisiopatológico en general. Este artículo describe evidencias recientes sobre las características de los autoanticuerpos anti-interferón alfa, y propone un acercamiento al conocimiento de su posible papel fisiopatológico en el desarrollo de las enfermedades de tipo autoinmune. Además, se discuten las bases moleculares de la diabetes mellitus tipo 1, y en especial, la asociación entre esta y las infecciones virales, que pueden desencadenar una respuesta local importante de producción de interferón alfa.
Palabras clave: diabetes mellitus tipo 1, autoanticuerpos, interferón alfa (INF-a), autoinmunidad.
ABSTRACT
Type 1 diabetes mellitus is an autoimmune disease characterized by destruction of the pancreatic beta cells due to a specific autoimmune response to these cells that is determined by the interaction between the genetic susceptibility and poorly defined environmental factors. Interferon alpha plays an important role as mediator of the inborn and the adaptive immune response of the body and its close relation with virus infection and onset of type 1 diabetes mellitus has been observed. The occurrence of natural to anti-interferon alpha auto-antibodies has called the attention of the international scientific community in the last few decades and this is an event present in roughly 10 % of patients with any autoimmune disease such as type 1 diabetes mellitus. However, the information on frequency, specificity, immunoreactivity and physiopathological role of this event is scanty in general. This article described recent evidence on the characteristics of anti-interferon alpha autoantibodies and approached to their possible physiopathological role in the development of autoimmune diseases. Additionally, it discussed the molecular bases of type 1 diabetes mellitus and particularly, the association between this disease and the viral infections, which may unleash a significant local response in terms of interferon alpha production.
Keywords: type 1 diabetes mellitus, autoantibodies, interferon alpha (INF-a), autoimmunity.
INTRODUCCIÓN
Una de las propiedades más importantes del sistema inmunológico de un individuo sano es su capacidad para reconocer y eliminar numerosos antígenos extraños y no responder perjudicialmente frente a moléculas propias, a esta ausencia de respuesta contra lo propio se le denomina tolerancia. La posibilidad de que este sistema reaccionara contra antígenos autólogos y produjera un daño tisular, fue observada por los inmunólogos desde el momento en que se describió su especificidad para reconocer a antígenos extraños.
Hoy se sabe que existen mecanismos y sistemas de regulación que, en determinadas circunstancias y bajo la influencia de diversos factores no bien dilucidados, se alteran y producen, en aquellos individuos susceptibles, fallos en los mecanismos de tolerancia, que una vez perpetuados, conllevan al desencadenamiento de enfermedades autoinmunes. Los autoantígenos frente a los cuales se levanta este tipo de respuesta autorreactiva nociva son de naturaleza diversa, y pueden ser hormonas, receptores, enzimas y citocinas;1 las citocinas, por su papel como mediadores de la respuesta inmunológica, reciben una importancia especial en este proceso.
Los interferones (INFs) son una familia de citocinas producidas por las células mononucleares como parte de la respuesta inmunológica contra virus y otros agentes infecciosos, ya que son capaces de inducir proteínas antivirales y activar a las células asesinas naturales (NK).2 Los tratamientos basados en INFs han sido usados para un grupo de condiciones patológicas, entre las que se encuentran hepatitis crónicas, leucemia y melanomas.3-5 Por otra parte, se ha reportado el desarrollo de enfermedades autoinmunes como efecto secundario del tratamiento con INFs, entre las que podemos encontrar la enfermedad de Behçet, el vitiligo, la tiroiditis autoinmune y la diabetes mellitus tipo 1 (DM 1).6-9
DESARROLLO
La DM 1, o diabetes autoinmune, es una enfermedad que se caracteriza por la destrucción de las células productoras de insulina del páncreas (células b), debido a una respuesta inmunológica específica contra ellas, que está determinada por la interacción entre la susceptibilidad genética (poligénica) y factores ambientales no bien definidos.10 Por su importancia y gravedad merece una atención especial, teniendo en cuenta que hace su aparición clínica mayoritariamente en edades tempranas de la vida. En Cuba, la prevalencia de la DM 1 en niños menores de 15 años es de 1,0 (tasa por 1 000 habitantes de estas edades).11 Según un estudio realizado en 7 países de América Latina y el Caribe por la OPS y la OMS, la tasa de prevalencia de diabetes diagnosticada entre adultos mayores entre 7 países (Uruguay, México, Cuba, Chile, Brasil, Barbados y Argentina) de América Latina y el Caribe es de 14,3 %. Las predicciones de la OMS indican que la prevalencia de la diabetes crecerá de 34 millones en el 2000 a 64 millones en 2025.12
Patogénesis de la DM 1
La característica principal de la DM 1 consiste en la destrucción selectiva de las células b, con la consecuente deficiencia de insulina. Esto ocurre en individuos en los que la susceptibilidad genética supera la protección genética, y es iniciada probablemente por factores ambientales que aún no han sido definidos con claridad. En la figura 1 se describe un modelo general de la destrucción de las células que conlleva al desarrollo de la DM 1.13 En la fase inicial la interacción entre los genes y los factores ambientales provocan una respuesta inmune con la aparición de autoanticuerpos como primera señal de la destrucción de las células b pancreáticas, seguida por la pérdida de la primera fase de la respuesta de la insulina. La progresión hacia el desarrollo de la diabetes resultante de una destrucción significativa de las células b es provocada por el desarrollo de un fenotipo de células T autoreactivas y un cambio del balance TH1/TH2 hacia un medio más pro-inflamatorio. La expresión del ligando Fas (FasL) sobre las células T citotóxicas, también marca la progresión hacia el establecimiento de la enfermedad.
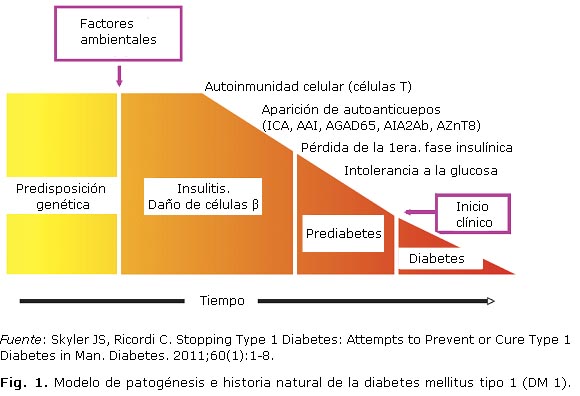
La DM 1 surge a través de un proceso inmune mediado por células, presumiblemente una reacción específica a una o más proteínas de las células b (autoantígenos).14 Como consecuencia se lleva a cabo un deterioro progresivo de la función de las células b y una disminución de su masa. Una respuesta inmune humoral secundaria es caracterizada por la aparición de autoanticuerpos que sirven como marcadores del daño inmunológico a las células b. Este es un proceso de la enfermedad que generalmente se desarrolla en un período que puede durar años, y además, puede ser variable como se observa en la figura 1. La disminución de la función y la masa de las células b se evidencia metabólicamente por la pérdida de la respuesta a la insulina en la primera fase después de una carga de glucosa intravenosa. El síndrome clínico de la DM 1 se hace evidente cuando se ha perdido la mayor parte de la función de las células b, y se presume que entre un 80 y un 90 % de su masa ha sido destruida en el momento de su diagnóstico,15 cuando acontece la hiperglicemia. Aunque esta es la secuencia general del desarrollo de la DM 1, aún quedan por dilucidar muchos aspectos en su patogénesis. Un mejor entendimiento de la naturaleza del proceso de la enfermedad, facilitaría el diseño de estrategias de intervención guiadas a frenar la destrucción de las células b y a la profilaxis de la DM 1.
Existen numerosos estudios que han evaluado la expresión de autoanticuerpos contra algunos componentes de las células de los islotes de Langerhans relacionados con la diabetes, tales como ICA, AAI, AGAD65, ICA512-IA216 y AZnT8,17 cuyos resultados sugieren que la aparición de estos autoanticuerpos constituye un factor de riesgo para el desarrollo futuro de la DM 1 en esos individuos. Sin embargo, aún no ha sido establecido en humanos el papel de los anticuerpos relacionados con diabetes en la patogénesis de esa enfermedad. De hecho, existe un caso reportado por la literatura científica que mostró el desarrollo de DM 1 en un paciente con agammaglobulinemia ligada a X, lo cual sugiere que en este caso los autoanticuerpos no fueron necesarios para la iniciación o progresión de la enfermedad.18
De manera general la DM 1 es considerada una enfermedad mediada por células T, y existen suficientes evidencias en ratones y humanos que lo sustentan.19 El examen de tejido pancreático obtenido por biopsia de pacientes con inicio reciente de DM 1 confirma insulitis con presencia de un infiltrado compuesto por linfocitos T CD4+ y T CD8+, linfocitos B y macrófagos,20 lo que sugiere que estas células desempeñan un papel importante en la destrucción de las células b pancreáticas.
Estudios preclínicos en ratones,21 y de manera más reciente estudios clínicos,22,23 han demostrado que el tratamiento con el anticuerpo monoclonal anti-CD3 puede prevenir la progresión de la DM 1 durante los primeros 2 años después del diagnóstico, a través de la reducción de la pérdida de la producción de insulina por las células b del páncreas. Aparentemente, en este mecanismo, está involucrada la inducción de tolerancia inmunológica, posiblemente mediada por células T reguladoras y sin la presencia de una supresión crónica inmune.
Los estudios hasta la fecha están enfocados en prolongar los efectos clínicos de la enfermedad, así como determinar el momento óptimo para la intervención. De sus resultados pudieran surgir nuevas estrategias guiadas a prevenir, o de manera más futurista, curar la DM 1.13,24
Diabetes inducida por virus
Existen diversos mecanismos por los cuales los virus pueden provocar daño a las células b. Los virus pueden infectar de manera directa y destruir las células b del páncreas, o pueden actuar de modo indirecto y desencadenar autoinmunidad con el consiguiente daño a esas células. Virus como el encefalomiocarditis (EMC-V) pueden inducir DM 1 por infección directa y destruir las células b por un mecanismo de lisis celular. Aunque este tipo de infección puede no ser suficiente para inducir insulitis o diabetes, la infección del tejido circundante puede provocar la destrucción de dichas células a través de la liberación de mediadores (citocinas) del sistema inmune.25 En contraste, los retrovirus pueden infectar células b e inducir la presentación de antígenos que permitan la autoinmunidad específica contra estas células.26 Por otra parte, virus como el coxsakievirus pueden infectar las células b e inducir la expresión de interferón alfa (INF-a), y como consecuencia, la expresión de otros mediadores que estimulan la llegada de linfocitos autorreactivos al páncreas para permitir la destrucción de las células b, y por tanto, la aparición de la enfermedad.27
La figura 2 muestra un panorama sobre los principales mecanismos involucrados en la patogénesis de la DM 1 inducida por virus. Entre las hipótesis más aceptadas se encuentra el mimetismo molecular entre epítopos de proteínas propias de agentes infecciosos y epítopos de autoantígenos.28 Tras una infección viral, podría darse una reacción cruzada, y generarse una respuesta contra epítopos de proteínas propias en los individuos susceptibles. La destrucción tisular y celular producida por una infección viral puede provocar la liberación de nuevos epítopos. Si algunos de estos epítopos fueran presentados y reconocidos por linfocitos T autorreactivos, estos podrían activarse y proliferar, e iniciar una respuesta autoinmunitaria. Otra hipótesis sugiere que la inflamación del órgano diana en el contexto de una infección sería el desencadenante del proceso autoinmunitario, inflamación que suele estar mediada por interferones de tipo 1, quimiocinas, citocinas pro-inflamatorias y otros factores de la inmunidad innata, que contribuirían a la activación de células T autorreactivas, de forma colateral e inespecífica.29 Otra posibilidad es la presentación de nuevos epítopos, procesados de una forma distinta después de una infección, o bien secuestrados hasta este momento en un órgano inmunoprivilegiado.30 Sin embargo, todos estos mecanismos son hipotéticos, ya que no se ha podido demostrar una relación causa-efecto de la enfermedad en humanos.
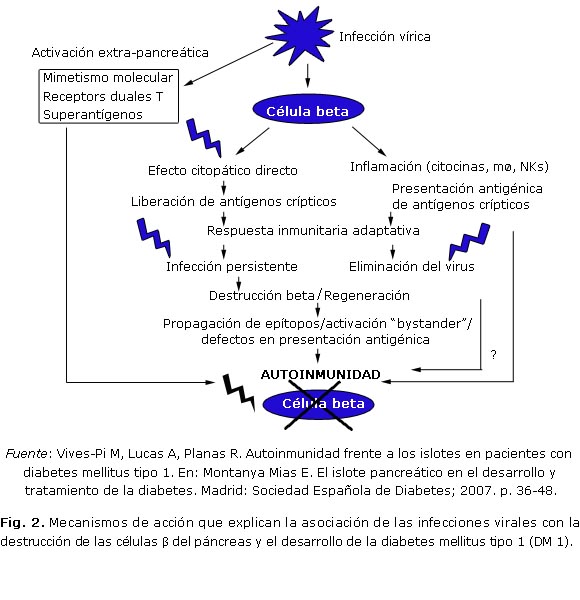
Hasta el momento, más de una decena de virus, con y sin tropismo por el páncreas, han sido asociados a la DM 1 en humanos y en modelos experimentales. Un ejemplo clásico es el virus de la rubéola, ya que se ha visto que niños con síndrome de la rubéola congénito presentan una mayor incidencia de diabetes.31 Probablemente, los virus que más se han relacionado con la DM 1 son los enterovirus y, dentro de este grupo, los echovirus y coxsackievirus A y B (especialmente el serotipo B4). Algunos estudios epidemiológicos sugieren una asociación entre infecciones por enterovirus y el posterior desarrollo de DM 1.32-34 Se han detectado anticuerpos y respuesta de linfocitos T contra enterovirus en pacientes diabéticos.35 Además, se reportó un caso en el que se pudo aislar un virus del páncreas de un paciente diabético en el inicio clínico de la enfermedad, y se trataba de una variante de coxsackievirus B4 (CVB4), que, además, era capaz de inducir la enfermedad en ratones.36 Los coxsackievirus son trópicos para las células b humanas, y entre los mecanismos que se proponen para el desarrollo de la enfermedad, se encuentran: la lisis directa de las células b e inducida por el virus, el mimetismo molecular, la activación inespecífica o bystander activation, y la persistencia del virus.37 Los rotavirus son los principales causantes de gastroenteritis durante la infancia, y se han relacionado también con DM 1. Las infecciones por rotavirus se han asociado a la aparición de autoanticuerpos específicos de islote. Además, se ha observado una similitud entre epítopos de rotavirus VP7 y autoantígenos de islote (GAD e IA-2).38 Estos son los ejemplos más evidentes, pero existen otros virus que también se han relacionado con la DM 1 en humanos y en modelos animales, como el citomegalovirus, el EMC-V, el mengovirus, los retrovirus endógenos y el virus Epstein-Barr.39
Sin embargo, aunque la base genética y los mecanismos anteriormente expuestos están involucrados en el aumento de la incidencia de la DM 1, ese incremento ha sido tan acelerado que cualquier explicación que omita los cambios medioambientales resultaría incompleta. La hipótesis de la higiene, recientemente reformulada como la hipótesis de los "viejos amigos", sugiere que desde el comienzo de la urbanización moderna ha existido un incremento progresivo de los problemas inmunorregulatorios atribuibles a la eliminación del medio ambiente urbano de organismos, también llamados "viejos amigos", con los cuales hemos coevolucionado, y cuya encomienda evolutiva fundamental ha sido precisamente el establecimiento de niveles basales "normales" de inmunorregulación.40 La pérdida de estos organismos, la dieta, los cambios en la microbiota intestinal, así como el retraso a la exposición a determinados virus en neonatos,41 son algunos de los factores que han provocado una disminución en la eficiencia de los mecanismos inmunorreguladores, y por tanto, han conllevado a un aumento de las enfermedades alérgicas, inflamatorias y de tipo autoinmune, como la DM 1.
Interferones
El término interferón fue usado por primera vez en 1957 por los investigadores Isaacs y Lindenmann42 para nombrar al factor soluble que producían las células al ser infectadas por un virus, que impedía la reinfección por otro virus, descubierto años antes por Nagano y Kojima.43 Más adelante se identificó a los INFs producidos por leucocitos (a) y por fibroblastos (b). El INF-g fue descubierto en 1965 por Wheelock, cuando encontró que cultivos de linfocitos incubados con fitohemaglutinina produjeron un agente con similitud al INF, inhibidor de los virus,44 y más recientemente fue descubierto un tercer grupo: la familia de INF-l.45
Los interferones pertenecen a una familia de citocinas constituida por glucoproteínas de bajo peso molecular, y pueden ser clasificados en 3 tipos antigénicamente diferentes, según la principal fuente celular que los produce y el estímulo necesario para su inducción: el tipo I (familia de INF-a, b), el tipo II (familia de INF-g) y un tercer grupo (familia de INF-l). Los interferones tipo I se dividen en 5 clases: INF-a, INF-b, INF-e, INF-k, y INF-w, en donde el INF-a y el INF-b son sus principales exponentes.46 El tipo II, por su parte, está representado por el INF-g, también llamado interferón inmune, que es sintetizado en el organismo por células del sistema inmune derivadas de células madres hematopoyéticas, entre las que se encuentran las células T colaboradoras (CD4+) y las células T citotóxicas (CD8+), que lo producen por estimulación con antígenos y las NK cuando son estimuladas por citocinas.47 El tipo III está constituido por 3 miembros que fueron denominados INF-l1, INF-l2 e INF-l3 o IL-28A, IL-28B e IL-29 respectivamente por 2 grupos de trabajo diferentes, y exhibe funciones similares a los INFs tipo I.48 Solo el grupo de los INF-a contiene subtipos muy relacionados entre sí, y presentan, a su vez, una homología del 60 % y 30 % con los INF-w e INF-b respectivamente.42 Los INF-a, b y w son expresados en niveles elevados por inducción mediada por infección viral, o a través de receptores de tipo Toll (TLR) en monocitos y células dendríticas plasmacitoides,49 y son importantes mediadores de la respuesta inmune innata. El interferón alfa 2b (INF-a2b), juega un importante papel, pues como se dijo anteriormente, se ha observado en estrecha relación con diversas enfermedades autoinmunes.
La señal natural más potente para la producción de INF tipo I es la infección viral. Experimentalmente la producción de INFs tipo I es estimulada por moléculas de ácido ribonucleico de doble cadena (ARNdc), que puede mimetizar una señal producida durante la replicación viral. Es secretado también durante la respuesta inmunitaria frente a los antígenos; en estos casos, las células T activadas por el antígeno estimulan a los fagocitos para que sinteticen INFs tipo I.33
INF-a: efectos biológicos y papel en la patogénesis de la DM 1
Los virus pudieran estar involucrados en la patogénesis de la DM 1 a través de la exposición de antígenos secuestrados en las células b dañadas, por alteración de algunos mecanismos de la tolerancia periférica, mímica molecular, o por la destrucción directa de las células productoras de insulina.18
Los INFs forman parte de la primera línea de defensa contra las infecciones virales. Los INFs tipo I, que incluyen al INF-a, tienen efectos antivirales, citostáticos e inmunomoduladores, que son de especial importancia en las infecciones virales. Diversos estudios indican que el INF-a actúa sobre las células dendríticas (CDs) para que estas maduren e induzcan eficientemente una respuesta de células T.50,51
Las CDs son cruciales en la unión existente entre el reconocimiento de los patógenos por el sistema inmune innato y la modulación de la respuesta inmune, principalmente debido a que las señales de los receptores asociados a la respuesta inmune innata pueden modificar su capacidad de presentación de antígenos y estimular a las células T respondedoras.52 De manera general la estimulación microbiana provoca que las CDs pasen de un estado inmaduro, en el cual estas incitan respuestas de células T tolerogénicas o abortivas, a un estado maduro, en el cual ellas generan una respuesta productiva. La maduración puede ser inducida directamente, debido a que las propias CDs expresan numerosos receptores, incluyendo algunos TLRs. Además, las CDs pueden responder indirectamente a la activación de los receptores asociados a la respuesta inmune innata por reconocimiento de señales, como pueden ser, las citocinas, producidas por otras células.53
Después de su interacción con el antígeno, las CDs migran a la zona de células T en los nódulos linfáticos donde interaccionan con las células T vírgenes. Durante la interacción con estas células, las CDs y bajo la influencia del microambiente anterior, tienen la capacidad de polarizar la respuesta de células T a un patrón TH1 o TH2. Los factores que influyen en este proceso son múltiples, y están relacionados con la presentación antigénica, su capacidad de producir citocinas polarizadoras como INF-a, IL-12, IL-6, la maduración de la red de CDs, el grado y tipo de coestimulación de las células T, y la presencia de inflamación durante el encuentro con el antígeno.54
Aunque gran diversidad de células pueden inducir la producción de INF-a, las CDs plasmocitoides humanas (células pre-dentríticas) son la mayor fuente de INF-a cuando existe exposición a virus.55 Sin embargo algunos experimentos refieren que las CDs plasmocitoides no son esenciales para la respuesta de INF-a contra determinados virus, lo que sugiere la existencia de otras fuentes alternativas de producción de INF-a in vivo.56
Además de la acción del INF-a sobre las CDs, esta citocina tiene numerosos efectos sobre el sistema inmune, como son, el aumento de la expresión de moléculas del complejo mayor de histocompatibilidad (MHC) clase I, la inhibición de la expresión de moléculas MHC clase II, la activación de células NK y la estimulación de la proliferación de células T CD8+.57 Sin embargo, la exposición prolongada del INF-a al sistema inmune puede romper la tolerancia e iniciar respuestas autoinmunes, que eventualmente pueden generar una enfermedad.58 Estudios recientes en pacientes con infección crónica por el virus de la hepatitis C, que fueron tratados con INF-a (Peg-INF-a/ribavirina), han mostrado una alta incidencia en el desarrollo de DM 1 durante o inmediatamente después de dicho tratamiento.59,60
Una expresión aumentada del INF-a, como marcador de infección viral, se correlaciona con diversas enfermedades autoinmunes, entre las que se incluye DM 1. La presencia de niveles elevados de INF-a en sangre de pacientes con DM 1, se ha visto relacionada con la infección por CVB4. Aparentemente el CVB4 estimula a las células b del páncreas a producir INF-a in vitro.61 La actividad inductora de INF-a del CVB4 es débil in vitro, pero se potencia in vivo con la interacción con IgG unidos a células y/o circulantes.62
Diversas investigaciones han mostrado que las células T juegan un papel fundamental en la patogénesis de la DM 1 durante las infecciones virales. Juhela S y otros, en el 2000 demostraron la presencia de una respuesta proliferativa al lisado de células infectadas por el coxsackievirus, que también incluía proteínas no estructurales del virus. La respuesta fue mayor en los niños que presentaban anticuerpos asociados a la DM 1 con respecto a los que no los presentaban. Esta diferencia fue más evidente en aquellos niños que poseían el alelo HLA-DQB1*02.63 Un estudio realizado recientemente en Cuba confirmó que existe asociación entre la presencia de la enfermedad y los alelos HLA-DQ y HLA-DR en la población cubana mestiza, en donde DQA1*0501, DQA1*0301, DQB1*0201 y DRB1*0301, DRB1*0401 son alelos de susceptibilidad, y DRB1*1501, DQA1*0102/3 y DQB1*0602 son alelos que confieren protección.64
A partir de estudios en animales se ha propuesto que el daño inicial de las células b infectadas por virus genera la producción de INF-a por estas células, y promueve un proceso inflamatorio y la autoinmunidad mediada por células.65 Otro estudio refiere que un intermediario replicativo de un ARNdc en combinación con INFs (INF-a o INF-b o INF-g), promueve la apoptosis de las células b. Una señalización excesiva a través de TLR e INF involucra efectos pro-apoptóticos y pro-inflamatorios, que potencialmente pueden llevar a una respuesta autoinmune en individuos predispuestos genéticamente. En este sentido, la expresión de INFs tipo I en células b de ratones diabéticos no obesos, rompe la tolerancia periférica de estas células y acelera la progresión de la diabetes.66 El posible papel de la infección viral en la muerte de las células b y la insulitis, se describen en la figura 2. Este modelo propone que un intermediario replicativo de un virus ARNdc se forma durante la infección viral, y estimula la expresión de TLR-3 y la señalización en las células b del páncreas. Esta activa al factor de transcripción nuclear kappa B (NF-kB) y coopera con el INF-g producido por las células del sistema inmune que invaden la zona, y se activa el trasductor de señal y activador de la traducción 1 (STAT-1). Tanto el NF-kB como el STAT-1 poseen propiedades pro-apoptóticas. El TLR-3 también promueve la inducción de INFs tipo I, el que a su vez, estimula la activación de STAT-1, la producción de quimiocinas y la sensibilización a la apoptosis. En las células b pancreáticas se producen quimiocinas que favorecen el reclutamiento de las células inmunes, lo que lleva a la exacerbación de la insulitis.67
Estas evidencias pueden indicar que una infección viral y las consecuencias que conlleva dicha infección, pueden estar relacionadas con el desarrollo de la diabetes. Una producción local de INF en las células b del páncreas debe ser suficientemente fuerte como para romper la tolerancia periférica, más que la central. Eldien HM y otros demostraron recientemente, en un modelo animal de DM 1, que bloqueando el receptor de INF tipo I se obtiene un aumento de la proliferación de los linfocitos en los órganos linfoides secundarios a través de: disminución de la apoptosis, modulacion de los niveles de glucosa en sangre y disminución de los niveles del factor de necrosis tumoral (TNF)-a. Se sugiere, entonces, que los niveles aumentados de INF-a durante la diabetes inducen una continua activación y agotamiento de los linfocitos, así como una alteración de su distribución en los órganos linfoides secundarios, sitio donde se produce el reconocimiento del antígeno y la respuesta inmune.68
La inducción de la autoinmunidad por INF-a es compatible con la detección de un transcriptoma de genes inducibles por esta citocina en los leucocitos de sangre periférica de pacientes con lupus eritematoso sistémico, junto a niveles elevados de esta molécula en el suero de estos pacientes.69 Este proceso parece estar mediado por la inducción de CXCL9, una quimiocina que atrae células autoinmunes que expresan CXCR-3, el receptor para CXCL9, y que media el ataque autoinmune contra las células b del páncreas. La estimulación de la expresión de MHC clase I por INF-a aumenta la susceptibilidad del ataque autoinmune de las células T CD8+, así como su capacidad citolítica. El INF-a puede contribuir a la retención de las células T CD8+ en los nódulos linfáticos a través de la estimulación de la expresión del marcador de activación CD69 en estas células, lo que llevaría a un mayor tiempo de exposición con las células presentadoras de antígenos.70
Por otra parte, la inducción de INF-g por INF-a ha sido reportada,71 y esto puede contribuir a un proceso de apoptosis de las células b del páncreas. La apoptosis de estas células pudiera permitir la exposición de nuevos antígenos, el procesamiento proteolítico de estos, y por lo tanto, una consecuente exposición de epítopos crípticos que pueden despertar la expansión de clones autoreactivos presentes en periferia. La propia molécula de INF-a puede ser un "blanco" de este proceso, y exponer nuevos epítopos que se convierten en autoantígenos, y por tanto, generan una respuesta de autoanticuerpos contra el INF-a.
Autoanticuerpos anti-INFa
La tolerancia a las proteínas propias es una característica básica del sistema inmune, y el desarrollo de anticuerpos contra proteínas autólogas es considerado como una ruptura de tolerancia. Aunque se pueden encontrar linfocitos B autorreactivos en individuos normales, usualmente estos no son estimulados a producir anticuerpos por los niveles circulantes de proteínas nativas.
Niveles bajos de autoanticuerpos de alta afinidad contra citocinas y factores de crecimiento, se han detectado en el suero de donantes sanos.72-74 Este tipo de autoanticuerpos son importantes efectores biológicos y moduladores de la respuesta inmune in vivo. Estos anticuerpos pueden inhibir la unión específica de las citocinas a sus receptores, e interferir con su actividad biológica. Se ha sugerido que juegan, además, un papel regulador en la intensidad y duración de la respuesta inmune.75
La caracterización de los autoanticuerpos contra INF-a indica que estos son de naturaleza policlonal, pertenecen a la clase de las inmunoglobulinas y muestran reactividad cruzada con diferentes subtipos de INFs tipo I.76 Varios reportes describen la presencia de autoanticuerpos contra citocinas como INF-a, INF-b, INF-g, interleucina (IL)1-a, IL-2, IL-6, IL-8, TNF-a y factores de crecimiento, tales como: factor de crecimiento de los nervios (NGF), factor estimulador de la formación de colonias de granulocitos y macrófagos (G-CSF, GM-CSF) en el suero de individuos sanos,68-70 pacientes con enfermedades autoinmunes,77,78 infecciones virales79 y tumores.80,81
No se conoce por qué estos anticuerpos se producen contra algunas citocinas, y no contra otras en personas aparentemente sanas. Sin embargo, es frecuente encontrar linfocitos B reactivos contra citocinas. Teniendo en cuenta que la cooperación de las células T es esencial en la producción de autoanticuerpos de tipo IgG de alta afinidad, la ausencia de tolerancia de células T parece estar involucrada en el curso de los eventos que llevan a la inducción de autoanticuerpos. Una posibilidad es que la activación por reactividad cruzada de linfocitos B y T se inicie si la citocina, como un hapteno, se uniera a un portador proporcionado por un patógeno.82 Por ejemplo, los poxvirus y herpes virus codifican para receptores de INF, así como TNF-a, IL-1-a e IL-6 receptores, que pueden funcionar como portadores inmunológicos.83
Una infección crónica o una rotura permanente de la tolerancia de células T contra epítopos en la molécula nativa de la citocina en cuestión, podrían explicar la producción continua de estos autoanticuerpos.78 Otra posibilidad es que la aparición inicial de linfocitos B productores de autoanticuerpos, se puede acelerar por la presencia del INF-a inducido por una infección, pero donde una generación significativa de linfocitos B y T autoinmunes solo se obtendría por una producción prolongada de este INF, que se mantendría como un mecanismo por círculo vicioso. Esta situación podría conllevar a la activación de CDs inmaduras, pero el INF-a endógeno sería producido de forma continua.33
Una expresión tejido-específica de antígenos en el contexto de la expresión de moléculas inmunorreguladoras inducidas por INF-a, también pueden estar en la base de este proceso inmunorregulador promovido por INF-a que caracteriza el desarrollo de la autoinmunidad. Se ha sugerido que el INF-a promueve la autoinmunidad por inducción de la expresión de superantígenos, y lleva a la activación de células T autoreactivas.84
Anticuerpos anti-INFa y DM 1
Un epítopo es un determinante antigénico o un sitio en la superficie de una molécula antigénica, al cual un solo anticuerpo se une. Algunos epítopos están ocultos (epítopos crípticos), ya que se encuentran localizados dentro de una región no expuesta de la molécula, y no están accesibles para ser reconocidos por los anticuerpos o los linfocitos T, hasta tanto se produzca un cambio conformacional o una alteración estereoquímica de la estructura. Al no poderse unir a los linfocitos, no tienen la posibilidad de inducir una respuesta inmune o tolerancia.85
Como ya se ha dicho, el INF-a inhibe la expresión de las moléculas de MHC clase II y activa a las células NK, y contribuyen al control y/o detención del reconocimiento de un autoantígeno y al establecimiento de células T autorreactivas. La presencia de autoanticuerpos contra INF-a en los procesos autoinmunes, puede estar relacionada con una condición patológica predeterminada que favorece el desarrollo de la enfermedad. Por otro lado, el tratamiento con INF-a2b promueve la aparición de enfermedades autoinmunes.6-8 La relación entre la presencia de los autoanticuerpos contra INF-a y el desarrollo de autoinmunidad no se ha establecido, como tampoco se ha esclarecido cómo la terapia prolongada con estas moléculas promueve la aparición de síntomas autoinmunes en los pacientes que reciben este tratamiento.
La aparición de autoanticuerpos contra INF-a en las enfermedades autoinmunes pudiera estar relacionada con la existencia de una producción de INF-a por las CDs, así como a un proceso de autoinmunización mediado por estas células. Ya que los autoanticuerpos han sido detectados solo en algunos pacientes, es probable que el proceso de formación de éstos, esté relacionado con factores genéticos.86
Las células b del páncreas pueden producir INF-a, presentar antígenos, y por tanto, conllevar a un proceso de autoinmunización, en el cual pueden participar eventos de mímica molecular con epítopos virales y/o de las células b. En este sentido es necesario subrayar que subtipos de receptores de acetilcolina (AChR) muestran cierto grado de homología estructural con zonas de la molécula de INF-a.82 Esta homología podría servir como fuente de reactividad cruzada y promover una autorreactividad contra la molécula de INF-a. Esta idea se soporta adicionalmente en que las células b del páncreas expresan en su superficie AChR.87 Además, los INFs (a, b, y w) son inducidos en niveles elevados por las CDs plasmacitoides, en monocitos, que pueden estar presentes en los nódulos linfáticos pancreáticos después de una infección viral o una estimulación a través de TLR.42 Este entorno pancreático de células productoras de INFs y presentadoras de antígenos, puede favorecer un proceso de autoinmunización del INF-a y provocar así la generación de anticuerpos contra esta molécula.
El proceso inflamatorio de la diabetes temprana se inicia y propaga por el efecto de las citocinas de tipo TH1 como los INFs, y es suprimida por las citocinas antiinflamatorias de tipo TH2. La estructura y función de las células b puede ser modulado por las citocinas Th1/Th2. La aparición de anticuerpos contra INF-a durante el proceso de desarrollo de la diabetes, pudiera formar parte de una respuesta para generar un balance entre la presencia de ciertos niveles de INF (umbral crítico), que define el inicio o no de una respuesta autoinmune contra las células b, o la protección frente a un ataque autoinmune. Sin embargo, el INF-a juega un rol bien complejo en la etiología de la DM 1, y la implicación del sistema INF en los principales mecanismos fisiopatológicos de esta enfermedad está aún por esclarecer.
REFERENCIAS BIBLIOGRÁFICAS
1.Abbas AK, Lichtman AH and Pillai S. Immunologic tolerance and autoimmunity. En: Abbas AK, Lichtman AH and Pillai S. Cellular and Molecular Immunology. 6th Updated Edition. Philadelphia: Elsevier-Masson; 2010. p. 319-44.
2. Sato K, Hida S, Takayanagi H, Yokochi T, Kayagaki N, Takeda K, et al. Anti-viral response by natural killer cells through TRAIL gene induction by interferon-alpha/beta. Eur J Immunol. 2001;31(11):3138-46.
3. Kawazoe T, Araki M, Lin Y, Ogawa M, Okamoto T, Yamamura T, et al. New-onset Type 1 diabetes mellitus and Anti-aquaporin-4 antibody positive optic neuritis associated with type 1 interferon therapy for chronic hepatitis C. Intern Med. 2012;51(18):2625-9.
4. Talpaz M, Hehlmann R, Quintás-Cardama A, Mercer J, Cortes J. Re-emergence of interferon-a in the treatment of chronic myeloid leukemia. Leukemia. 2013;27(4):803-12.
5. Rozati S, Naef L, Levesque MP, French LE, Dummer R. Real-life Experience With Pegylated Interferon and Conventional Interferon in Adjuvant Melanoma Therapy. J Immunother. 2013;36(1):52-6.
6. Monastirli A, Chroni E, Georgiou S, Ellul J, Pasmatzi E, Papathanasopoulos P, et al. Interferon-a treatment for acute myelitis and intestinal involvement in severe Behçet's disease. QJM. 2010;103(10):787-90.
7. Guney E, Akcali G, Akcay BI, Unlu C, Erdogan G, Bozkurt TK, et al. Vitiligo in a Patient Treated with Interferon Alpha-2a for Behçet's Disease. Case Report Med [serie en Internet]. 2012 Jul [citado 12 de enero de 2013];(2012). Disponible en: http://www.hindawi.com/crim/medicine/2012/387140/
8. Kose S, Gozaydin A, Akkoclu G, Ece G. Chronic hepatitis B with type I diabetes mellitus and autoimmune thyroiditis development during interferon alpha therapy. J Infect Dev Ctries. 2012;6(4):364-8.
9. Nakamura K, Kawasaki E, Abiru N, Jo O, Fukushima K, Satoh T, et al. Trajectories of ant-islet autoantibodies before development of type 1 diabetes in interferon-treated hepatitis C patients. Case reports and a literature review. Endocr J. 2010;57(11):947-51.
10. Von Herrath M. Diabetes: a virus-gene collaboration. Nature. 2009;459(7246):518-9.
11. Ministerio de Salud Pública, Direccion Nacional de Estadística. Anuario Estadístico del Registro Nacional de Dispensarización. MINSAP (Cuba); 2009.
12. Barceló A, Pelaez M, Rodríguez-Wong L, Meiners M. Prevalencia de la diabetes entre adultos mayores en siete países de América Latina y el Caribe (ALC): Proyecto SABE (Salud, Bienestar y Envejecimiento). Washington DC: Organización Panamericana de la Salud/Organización Mundial de la Salud (OPS/OMS); 2004.
13. Skyler JS, Ricordi C. Stopping type 1 diabetes: attempts to prevent or cure type 1 diabetes in man. Diabetes. 2011;60(1):1-8.
14. Eisenbarth GS. Banting Lecture 2009: an unfinished journey: molecular pathogenesis to prevention of type 1 diabetes. Diabetes. 2010;59(4):759-74.
15. Jerry P. Palmer. C-peptide in the natural history of type 1 diabetes. Diabetes Metab Res Rev. 2009;25(4):325-8.
16. Wenzlau JM, Moua O, Sarkar SA, Yu L, Rewers M, Eisenbarth GS, et al. SlC30A8 is a major target of humoral autoimmunity in type 1 diabetes and a predictive marker in prediabetes. Ann NY Acad Sci. 2008;1150:256-9.
17. Winter WE, Schatz DA. Autoimmune markers in diabetes. Clin Chem. 2011;57(2):168-75.
18. Martin S, Wolf-Eichbaum D, Duinkerken G, Scherbaum WA, Kolb H, Noordzij JG, et al. Development of type 1 diabetes despite severe hereditary B lymphocyte deficiency. N Engl J Med. 2001;345(14):1036-45.
19. Coppieters KT, Boettler T, von Herrath M. Virus Infections in Type 1 Diabetes. Cold Spring Harb Perspect Med. 2012;2(1):a007682.
20. Imagawa A, Hanafusa T, Itoh N, Waguri M, Yamamoto K, Miyagawa J, et al. Immunological abnormalities in islet at diagnosis paralleled further deterioration of glycaemic control in patients with recent-oncet type 1 diabetes mellitus. Diabetologia. 1999;42(5):574-8.
21. Sherry NA, Chen W, Kushner JA, Glandt M, Tang Q, Tsai S, et al. Exendin-4 improves reversal of diabetes in NOD mice treated with anti-CD3 monoclonal antibody by enhancing recovery of beta-cells. Endocrinology. 2007;148(11):5136-44.
22. Kaufman A, Herold KC. Anti-CD3 mAbs for treatment of type 1 diabetes. Diabetes Metab Res Rev. 2009;25(4):302-6.
23. Michels AW, Eisenbarth GS. Immune intervention in type 1 diabetes. Semin Immunol. 2011;23(3):214-9.
24. Ablamunits V, Henegariu O, Hansen JB, Opare-Addo L, Preston-Hurlburt P, Santamaria P, et al. Synergistic reversal of type 1 diabetes in NOD mice with anti-CD3 and interleukin-1 blockade: evidence of improved immune regulation. Diabetes. 2012;61(1):145-54.
25. Rose NR. Autoimmunity in coxsackievirus infection. Curr Top Microbiol Immunol. 2008;323:293-314.
26. Marquerat S, Wang WY, Todd JA, Conrad B. Association of human endogenous retrovirus K-18 polymorphisms with type 1 diabetes. Diabetes. 2004;53(3):852-4.
27. Huber S. Host immune responses to coxsackievirus B3. Curr Top Microbiol Immunol. 2008;323:199-221.
28. Coppieters KT, Wiberg A, von Herrath MG. Viral infections and molecular mimicry in type 1 diabetes. APMIS. 2012;120(12):941-9.
29. Csorba TR, Lyon AW, Hollenberg MD. Autoimmunity and the pathogenesis of type 1 diabetes. Crit Rev Clin Lab Sci. 2010;47(2):51-71.
30. Hober D, Sauter P. Pathogenesis of type 1 diabetes mellitus: interplay between enterovirus and host. Nat Rev Endocrinol. 2010;6(5):279-89.
31. McEvoy RC, Fedun B, Cooper LZ, Thomas NM, Rodriguez de Cordoba S, Rubinstein P, et al. Children at High Risk of diabetes mellitus: New York studies of families with diabetes and of children with congenital Rubeolla syndrome. Adv Exp Med Biol. 1988;246:221-7.
32. Sarmiento L, Cabrera-Rode E, Leukeni L, Cuba I, Molina G, Fonseca M, et al. Occurrence of enterovirus RNA in serum of children with newly diagnosed type 1 diabetes and islet cell autoantibody-positive subjects in a population with a low incidence of type 1 diabetes. Autoimmunity. 2007;40(7):540-5.
33. Oikarinen S, Martiskainen M, Tauriainen S, Huhtala H, Ilonen J, Veijola R, et al. Enterovirus RNA in blood is linked to the development of type 1diabetes. Diabetes. 2011;60(1):276-9.
34. Sarmiento L, Cubas-Dueñas I, Cabrera-Rode E. Evidence of association between Type 1 Diabetes and exposure to Enterovirus in Cuban children and adolescents. MEDICC Rev. 2013;15(1):29-32.
35. Schulte BM, Lanke KH, Piganelli JD, Kers-Rebel ED, Bottino R, Trucco M, et al. Cytokine and Chemokine Production by Human Pancreatic Islets Upon Enterovirus Infection. Diabetes. 2012;61(8):2030-6.
36. Honeyman M. How robust is the evidence for viruses in the induction of type 1 diabetes? Curr Opin Immunol. 2005;17(6):616-23.
37. Jaïdane H, Hober D. Role of Coxsackievirus B4 in the pathogenesis of type 1 diabetes. Diabetes Metab. 2008;34(6 Pt 1):537-48.
38. Honeyman MC, Stone NL, Harrison LC. T-cell epitopes in type 1 diabetes autoantigen tyrosine phosphatase IA-2: potential for mimicry with rotavirus and other environmental agents. Mol Med. 1998;4(4):231-9.
39. Loechelt BJ, Boulware D, Green M, Baden LR, Gottlieb P, Krause-Steinrauf H, et al. Epstein-Barr and Other Herpesvirus Infections in Patients With Early Onset Type 1 Diabetes Treated With Daclizumab and Mycophenolate Mofetil. Clin Infect Dis. 2013;56(2):248-54.
40. Rook GA. Hygiene hypothesis and autoimmune diseases. Clin Rev Allergy Immunol. 2012;42(1):5-15.
41. Tracy S, Drescher KM, Jackson JD, Kim K, Kono K. Enteroviruses, type 1 diabetes and hygiene: a complex relationship. Rev Med Virol. 2010;20(2):106-16.
42. Isaacs A, Lindenmann J. Virus Interference. I. The interferon. Proc R Soc Lond B Biol Sci. 1957;147(927):258-67.
43. Nagano Y, Kojima Y. Immunizing property of vaccinia virus inactivated by ultraviolets rays (Article in French). CR Seances Soc Biol Fil. 1954;148(19-20):1700-2.
44. Wheelock EF. Interferon-like virus inhibitor induced in human leukocytes by phytohemaglutinin. Science. 1965;149(3681):310-1.
45. Vilcek J. Novel Interferons. Nature Immunology. 2004;4(1):8-9.
46. Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8-32.
47. Tian Z, Gershwin ME, Zhang C. Regulatory NK cells in autoimmune disease. J Autoimmun. 2012;39(3):206-15.
48. Charles E. Interferons, Interferon Receptors, Signal Transducer and Transcriptional Activators, and Interferon Regulatory Factors. J Biol Chem. 2007;282(28):20045-6.
49. Hertzog PJ, O'Neill LA, Hamilton JA. The interferon in TLR signalling: more than just antiviral. Trends Immunol. 2003;24(10):534-9.
50. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245-52.
51. Mayer CT, Berod L, Sparwasser T. Layers of dendritic cell-mediated T cell tolerance, the irregulation and the prevention of autoimmunity. Front Immunol. 2012;3:183.
52. Manicassamy S, Pulendran B. Dendritic cell control of tolerogenic responses. Immunological Reviews. 2011;241(1):206-27.
53. Tisch R. Immunogenic versus Tolerogenic Dendritic Cells: A Matter of Maturation. Int Rev Immunol. 2010;29(2):111-8.
54. Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol. 2006;311:17-58.
55. Gottenberg JE, Chiocchia G. Dendritic cells and interferon-mediated autoimmunity. Biochimie. 2007;89(6-7):856-71.
56. Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Brière F, et al. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J Exp Med. 2002;195(4):517-28.
57. Taki S. Type I interferons and autoimmunity: lessons from the clinic and from IRF-2-deficient mice. Cytokine Growth Factor Rev. 2002;13(4-5):379-91.
58. Kawazoe T, Araki M, Lin Y, Ogawa M, Okamoto T, Yamamura T, et al. New-onset type 1 diabetes mellitus and anti-aquaporin-4 antibody positive optic neuritis associated with type 1 interferon therapy for chronic hepatitis C. Intern Med. 2012;51(18):2625-9.
59. Schreuder TC, Gelderblom HC, Weegink CJ, Hamann D, Reesink HW, Devries JH, et al. High incidence of type 1 diabetes mellitus during or shortly after treatment with pegylated interferon alpha for chronic hepatitis C virus infection. Liver Int. 2008;28(1):39-46.
60. Vinagre I. Aparición de diabetes mellitus tipo 1 durante el tratamiento con interferón alfa: dos casos clínicos. Endocrinol Nutr. 2010;57:393-5.
61. Chehadeh W, Kerr-Conte F, Pattou F, Alm G, Lefebvre J, Wattré P, et al. Persistent infection of human pancreatic islets by coxsakievirus B is associated with alpha interferon synthesis in beta cells. J Virol. 2000;74(21):10153-64.
62. Hober D, Chehadeh W, Weill J, Hober C, Vantyghem MC, Gronnier P, et al. Circulating and cell-bound antibodies increase coxsackievirus B4-induced production of IFN-alpha by peripheral blood mononuclear cells from patients with type 1 diabetes. J Gen Virol. 2002;83(Pt 9):2169-76.
63. Juhela S, Hyoty H, Roivainen M, Härkönen T, Putto-Laurila A, Simell O, et al. T-cell responses to enterovirus antigens in children with type 1 diabetes. Diabetes. 2000;49(8):1308-13.
64. Díaz-Horta O, Cintado A, Fernández-De-Cossio ME, Nazabal M, Ferrer A, Roca J, et al. Relationship of Type 1 diabetes to ancestral proportions and HLA DR/DQ alleles in a sample of the admixed Cuban population. Ann Hum Biol. 2010;37(6):778-88.
65. Alba A, Puertas MC, Carrillo J, Planas R, Ampudia R, Pastor X, et al. IFN beta Accelerates Autoimmune Type 1 diabetes in NOD Mice and breaks the tolerance to cells in nondiabetes-prone mice 1. J Immunol. 2004;173(11):6667-75.
66. Li Q, McDevitt H. The role of interferon alpha in initiation of type I diabetes in the NOD mouse. Clin Immunol. 2011;140(1):3-7.
67. Rasschaert J, Ladriere L, Urbain Z, Dogusan Z, Katabua B, Sato S, et al. Toll-like Receptor 3 and STAT-1 Contribute to Double-stranded RNA+ Interferon-gamma-induced Apoptosis in Primary Pancreatic beta-Cells. J Biol Chem. 2005;280(40):33984-91.
68. Eldien HM, El-Elaimy IA, Ibrahim HM, Badr BM, Rabah DM, Badr G, et al. Increased level of type 1 Interferon (IFN) during type 1 diabetes (T1D) induces apoptosis in spleen-homing T cells. Afric J Pharma Pharmac. 2012;6(37):2675-81.
69. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. 2003;100(5):2610-5.
70. Colonna M. Toll-like receptors and IFN-alpha: partners in autoimmunity. J Clin Invest. 2006;116(9):2319-22.
71. Zeestraten EC, Speetjens FM, Welters MJ, Saadatmand S, Stynenbosch LF, Jongen R, et al. Addition of interferon-alpha to the p53-SLP(®) vaccine results in increased production of interferon-gamma in vaccinated colorectal cancer patients: a phase I/II clinical trial. Int J Cancer. 2013;132(7):1581-91.
72. Hansen MB, Svenson M, Diamant M, Bendtzen K. Anti-interleukin 6 antibodies in normal human serum. Scand J Immunol. 1991;33(6):777-81.
73. Tiberio L, Caruso A, Pozzi A, Rivoltini L, Morelli D, Monti E, et al. The detection and biological activity of human antibodies to IL-2 in normal donors. Scand J Immunol. 1993;38(5):472-6.
74. Ibrahim EH. Anti-IFN autoantibodies are present in healthy Egyptian blood donors at low titer. Cell Immunol. 2011;271(2):365-70.
75. van der Meide PH, Schellekens H. Anticytokine autoantibodies: epiphenomenon or critical modulators of cytokine action. Biotherapy. 1997;10(1):39-48.
76. Prummer O, Seyfarth C, Scherbaum W, Drees N, Porzsolt F. Interferon alpha antibodies in auto-immune diseases. J Interferon Res. 1989;9 Suppl 1:S567-74.
77. Dicou E, Perrot S, Menkes CJ, Masson C, Nerriere V. Nerve growth factor (NGF) autoantibodies and NGF in the synovial fluid: Implications in spondylarthropaties. Autoimmunity. 1996;24(1):1-9.
78. Trotter JL, Damico CA, Trotter AL, Collins KG, Cross AH. Interleukin-2 binding proteins in sera from normal subjects and multiple sclerosis patients. Neurology. 1995;45(11):1971-4.
79. Caruso A, Bonfanti C, Colombrita D, De Francesco M, De Rango C, Foresti I, et al. Natural antibodies to IFN gamma in man and their increase during viral infection. J Immunol. 1990;144(2):685-90.
80. Laricchia-Robbio L, Moscato S, Genua A, Liberati AM, Revoltella RP. Naturally occurring and therapy-induced antibodies to human granulocyte colony-stimulating factor (G-CSF) in human serum. J Cell Physiol. 1997;173(2):219-26.
81. Burbelo PD, Browne SK, Sampaio EP, Giaccone G, Zaman R, Kristosturyan E, et al. Anti-cytokine autoantibodies are associated with opportunistic infection in patients with thymic neoplasia. Blood. 2010;116(23):4848-58.
82. Bendtzen K, Hansen MB, Ross C, Svenson M. High-avidity autoantibodies to cytokines. Immunol Today. 1998;19(5):209-11.
83. Spriggs MK. One step ahead of the game: viral immunomodulatory molecules. Annu Rev Immunol. 1996;14:101-30.
84. Stauffer Y, Marguerat S, Meylan F, Ucla C, Sutkowski N, Huber B, et al. Interferon-alpha-induced endogenous superantigen a model linking environment and autoimmunity. Immunol. 2001;15(4):591-601.
85. Powell AM and Black MM. Epitope spreading: protection from pathogen, but propagation of autoimmunity? Clin Exp Dermatol. 2001;26(5):427-33.
86. Bello-Rivero I, Cervantes M, Torres Y, Ferrero J, Rodríguez E, Pérez J, et al. Characterization of the Immunoreactivity of AntiInterferon Alpha Antibodies in Myasthenia Gravis Patients. Epitope mapping. J Autoimmun. 2004;23(1):63-73.
87. Verchere CB. Modulation of acetylcholine-stimulated insulin release by glucose and gastric inhibitory polypeptide. Pharmacol. 1991;42(5):273-82.
Recibido: 27 de enero de 2013.
Aprobado: 12 de febrero de 2013.
Alieski Cruz Ramírez. Centro de Ingeniería Genética y Biotecnología (CIGB). Avenida 31 entre 156 y 190, Reparto Cubanacán, municipio Playa. La Habana, Cuba. Correo electrónico: alieski.cruz@cigb.edu.cu
